General Information About Non-Hodgkin Lymphoma (NHL)
The non-Hodgkin lymphomas (NHL) are a heterogeneous group of lymphoproliferative malignancies with differing patterns of behavior and responses to treatment.[1]
Like Hodgkin lymphoma, NHL usually originates in lymphoid tissues and can spread to other organs. NHL, however, is much less predictable than Hodgkin lymphoma and has a far greater predilection to disseminate to extranodal sites. The prognosis depends on the histological type, stage, and treatment.
Incidence and Mortality
Estimated new cases and deaths from NHL in the United States in 2023:[2]
- New cases: 80,550.
- Deaths: 20,180.
Anatomy
NHL usually originates in lymphoid tissues.
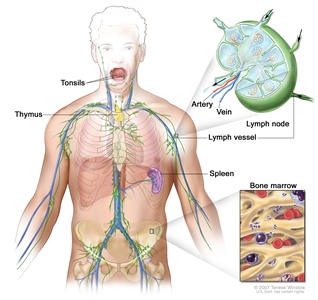
Anatomy of the lymph system.
Prognosis and Survival
NHL can be divided into two prognostic groups: the indolent lymphomas and the aggressive lymphomas.
Indolent NHL types have a relatively good prognosis, with a median survival as long as 20 years, but they usually are not curable in advanced clinical stages.[3] Early-stage (stage I and stage II) indolent NHL can be effectively treated with radiation therapy alone. Most of the indolent types are nodular (or follicular) in morphology.
The aggressive type of NHL has a shorter natural history, but a significant number of these patients can be cured with intensive combination chemotherapy regimens.
In general, with modern treatment of patients with NHL, the overall survival rate at 5 years is over 60%. More than 50% of patients with aggressive NHL can be cured. Most relapses occur in the first 2 years after therapy. The risk of late relapse is higher in patients who manifest both indolent and aggressive histologies.[4]
While indolent NHL is responsive to immunotherapy, radiation therapy, and chemotherapy, a continuous rate of relapse is usually seen in advanced stages. However, patients can often be re-treated with considerable success if the disease histology remains low grade. Patients who present with or convert to aggressive forms of NHL may have sustained complete remissions with combination chemotherapy regimens or aggressive consolidation with marrow or stem cell support.[5,6]
References:
- Shankland KR, Armitage JO, Hancock BW: Non-Hodgkin lymphoma. Lancet 380 (9844): 848-57, 2012.
- American Cancer Society: Cancer Facts and Figures 2023. American Cancer Society, 2023. Available online. Last accessed January 13, 2023.
- Tan D, Horning SJ, Hoppe RT, et al.: Improvements in observed and relative survival in follicular grade 1-2 lymphoma during 4 decades: the Stanford University experience. Blood 122 (6): 981-7, 2013.
- Cabanillas F, Velasquez WS, Hagemeister FB, et al.: Clinical, biologic, and histologic features of late relapses in diffuse large cell lymphoma. Blood 79 (4): 1024-8, 1992.
- Bastion Y, Sebban C, Berger F, et al.: Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol 15 (4): 1587-94, 1997.
- Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995.
Late Effects of Treatment of NHL
Late effects of treatment of non-Hodgkin lymphoma (NHL) have been observed. Impaired fertility may occur after exposure to alkylating agents.[1] For as many as three decades after diagnosis, patients are at a significantly elevated risk of developing second primary cancers, especially the following:[2,3,4,5]
- Lung cancer.
- Brain cancer.
- Kidney cancer.
- Bladder cancer.
- Melanoma.
- Hodgkin lymphoma.
- Acute nonlymphocytic leukemia.
Left ventricular dysfunction was a significant late effect in long-term survivors of high-grade NHL who received more than 200 mg/m² of doxorubicin.[1,6]
Myelodysplastic syndrome and acute myelogenous leukemia are late complications of myeloablative therapy with autologous bone marrow or peripheral blood stem cell support, as well as conventional chemotherapy-containing alkylating agents.[3,7,8,9,10,11,12,13,14] Most of these patients show clonal hematopoiesis even before the transplantation, suggesting that the hematologic injury usually occurs during induction or reinduction chemotherapy.[9,15,16] With a median 10-year follow-up after autologous bone marrow transplantation (BMT) with conditioning using cyclophosphamide and total-body radiation therapy, the incidence of a second malignancy was 21% in a series of 605 patients, and 10% of those were solid tumors.[17]
Successful pregnancies with children born free of congenital abnormalities have been reported in young women after autologous BMT.[18] Late-occurring venous thromboembolism can occur after allogeneic or autologous BMT.[19]
Long-term impaired immune health was evaluated in a retrospective cohort study of 21,690 survivors of diffuse large B-cell lymphoma from the California Cancer Registry. Elevated incidence rate ratios were found up to 10 years later for pneumonia (10.8-fold), meningitis (5.3-fold), immunoglobulin deficiency (17.6-fold), and autoimmune cytopenias (12-fold).[20] Similarly, there are impaired humoral responses to COVID-19 virus vaccination in patients with lymphoma who receive B-cell–directed therapies.[21,22]
Some patients have osteopenia or osteoporosis at the start of therapy; bone density may worsen after therapy for lymphoma.[23]
References:
- Haddy TB, Adde MA, McCalla J, et al.: Late effects in long-term survivors of high-grade non-Hodgkin's lymphomas. J Clin Oncol 16 (6): 2070-9, 1998.
- Travis LB, Curtis RE, Glimelius B, et al.: Second cancers among long-term survivors of non-Hodgkin's lymphoma. J Natl Cancer Inst 85 (23): 1932-7, 1993.
- Mudie NY, Swerdlow AJ, Higgins CD, et al.: Risk of second malignancy after non-Hodgkin's lymphoma: a British Cohort Study. J Clin Oncol 24 (10): 1568-74, 2006.
- Hemminki K, Lenner P, Sundquist J, et al.: Risk of subsequent solid tumors after non-Hodgkin's lymphoma: effect of diagnostic age and time since diagnosis. J Clin Oncol 26 (11): 1850-7, 2008.
- Major A, Smith DE, Ghosh D, et al.: Risk and subtypes of secondary primary malignancies in diffuse large B-cell lymphoma survivors change over time based on stage at diagnosis. Cancer 126 (1): 189-201, 2020.
- Moser EC, Noordijk EM, van Leeuwen FE, et al.: Long-term risk of cardiovascular disease after treatment for aggressive non-Hodgkin lymphoma. Blood 107 (7): 2912-9, 2006.
- Darrington DL, Vose JM, Anderson JR, et al.: Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol 12 (12): 2527-34, 1994.
- Stone RM, Neuberg D, Soiffer R, et al.: Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 12 (12): 2535-42, 1994.
- Armitage JO, Carbone PP, Connors JM, et al.: Treatment-related myelodysplasia and acute leukemia in non-Hodgkin's lymphoma patients. J Clin Oncol 21 (5): 897-906, 2003.
- André M, Mounier N, Leleu X, et al.: Second cancers and late toxicities after treatment of aggressive non-Hodgkin lymphoma with the ACVBP regimen: a GELA cohort study on 2837 patients. Blood 103 (4): 1222-8, 2004.
- Oddou S, Vey N, Viens P, et al.: Second neoplasms following high-dose chemotherapy and autologous stem cell transplantation for malignant lymphomas: a report of six cases in a cohort of 171 patients from a single institution. Leuk Lymphoma 31 (1-2): 187-94, 1998.
- Lenz G, Dreyling M, Schiegnitz E, et al.: Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol 22 (24): 4926-33, 2004.
- McLaughlin P, Estey E, Glassman A, et al.: Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 105 (12): 4573-5, 2005.
- Morton LM, Curtis RE, Linet MS, et al.: Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 28 (33): 4935-44, 2010.
- Mach-Pascual S, Legare RD, Lu D, et al.: Predictive value of clonality assays in patients with non-Hodgkin's lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91 (12): 4496-503, 1998.
- Lillington DM, Micallef IN, Carpenter E, et al.: Detection of chromosome abnormalities pre-high-dose treatment in patients developing therapy-related myelodysplasia and secondary acute myelogenous leukemia after treatment for non-Hodgkin's lymphoma. J Clin Oncol 19 (9): 2472-81, 2001.
- Brown JR, Yeckes H, Friedberg JW, et al.: Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 23 (10): 2208-14, 2005.
- Jackson GH, Wood A, Taylor PR, et al.: Early high dose chemotherapy intensification with autologous bone marrow transplantation in lymphoma associated with retention of fertility and normal pregnancies in females. Scotland and Newcastle Lymphoma Group, UK. Leuk Lymphoma 28 (1-2): 127-32, 1997.
- Gangaraju R, Chen Y, Hageman L, et al.: Risk of venous thromboembolism in patients with non-Hodgkin lymphoma surviving blood or marrow transplantation. Cancer 125 (24): 4498-4508, 2019.
- Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020.
- Ghione P, Gu JJ, Attwood K, et al.: Impaired humoral responses to COVID-19 vaccination in patients with lymphoma receiving B-cell-directed therapies. Blood 138 (9): 811-814, 2021.
- Terpos E, Trougakos IP, Gavriatopoulou M, et al.: Low neutralizing antibody responses against SARS-CoV-2 in older patients with myeloma after the first BNT162b2 vaccine dose. Blood 137 (26): 3674-3676, 2021.
- Westin JR, Thompson MA, Cataldo VD, et al.: Zoledronic acid for prevention of bone loss in patients receiving primary therapy for lymphomas: a prospective, randomized controlled phase III trial. Clin Lymphoma Myeloma Leuk 13 (2): 99-105, 2013.
Cellular Classification of NHL
A pathologist should be considered for consultation before a biopsy because some studies require special preparation of tissue (e.g., frozen tissue). Knowledge of cell surface markers and immunoglobulin and T-cell receptor gene rearrangements may help with diagnostic and therapeutic decisions. The clonal excess of light-chain immunoglobulin may differentiate malignant from reactive cells. Since the prognosis and the approach to treatment are influenced by histopathology, outside biopsy specimens should be carefully reviewed by a hematopathologist who is experienced in diagnosing lymphomas. Although lymph node biopsies are recommended whenever possible, sometimes immunophenotypic data are sufficient to allow diagnosis of lymphoma when fine-needle aspiration cytology is preferred.[1,2]
Historical Classification Systems
Historically, uniform treatment of patients with non-Hodgkin lymphoma (NHL) has been hampered by the lack of a uniform classification system. In 1982, results of a consensus study were published as the Working Formulation.[3] The Working Formulation combined results from six major classification systems into one classification. This allowed comparison of studies from different institutions and countries. The Rappaport classification, which also follows, is no longer in common use.
Table 1. Historical Classification Systems for Non-Hodgkin Lymphoma (NHL)| Working Formulation[3] | Rappaport Classification |
|---|
| Low grade | |
| A. Small lymphocytic, consistent with chronic lymphocytic leukemia | Diffuse lymphocytic, well-differentiated |
| B. Follicular, predominantly small-cleaved cell | Nodular lymphocytic, poorly differentiated |
| C. Follicular, mixed small-cleaved, and large cell | Nodular mixed, lymphocytic, and histiocytic |
| Intermediate grade | |
| D. Follicular, predominantly large cell | Nodular histiocytic |
| E. Diffuse, small-cleaved cell | Diffuse lymphocytic, poorly differentiated |
| F. Diffuse mixed, small and large cell | Diffuse mixed, lymphocytic, and histiocytic |
| G. Diffuse, large cell, cleaved, or noncleaved cell | Diffuse histiocytic |
| High grade | |
| H. Immunoblastic, large cell | Diffuse histiocytic |
| I. Lymphoblastic, convoluted, or nonconvoluted cell | Diffuse lymphoblastic |
| J. Small noncleaved-cell, Burkitt, or non-Burkitt | Diffuse undifferentiated Burkitt or non-Burkitt |
Current Classification Systems
As the understanding of NHL has improved and as the histopathological diagnosis of NHL has become more sophisticated with the use of immunologic and genetic techniques, a number of new pathological entities have been described.[4] In addition, the understanding and treatment of many of the previously described pathological subtypes have changed. As a result, the Working Formulation has become outdated and less useful to clinicians and pathologists. Thus, European and American pathologists have proposed a new classification, the Revised European American Lymphoma (REAL) classification.[5,6,7,8] Since 1995, members of the European and American Hematopathology societies have been collaborating on a new World Health Organization (WHO) classification, which represents an updated version of the REAL system.[9,10]
The WHO modification of the REAL classification recognizes three major categories of lymphoid malignancies based on morphology and cell lineage: B-cell neoplasms, T-cell/natural killer (NK)-cell neoplasms, and Hodgkin lymphoma (HL). Both lymphomas and lymphoid leukemias are included in this classification because both solid and circulating phases are present in many lymphoid neoplasms and distinction between them is artificial. For example, B-cell chronic lymphocytic leukemia (CLL) and B-cell small lymphocytic lymphoma are simply different manifestations of the same neoplasm, as are lymphoblastic lymphomas and acute lymphocytic leukemias. Within the B-cell and T-cell categories, two subdivisions are recognized: precursor neoplasms, which correspond to the earliest stages of differentiation, and more mature differentiated neoplasms.[9,10]
Updated REAL/WHO classification
B-cell neoplasms
- Precursor B-cell neoplasm: precursor B-acute lymphoblastic leukemia/lymphoblastic lymphoma (LBL).
- Peripheral B-cell neoplasms.
- B-cell CLL/small lymphocytic lymphoma.
- B-cell prolymphocytic leukemia.
- Lymphoplasmacytic lymphoma/immunocytoma.
- Mantle cell lymphoma.
- Follicular lymphoma.
- Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphatic tissue (MALT) type.
- Nodal marginal zone B-cell lymphoma (± monocytoid B-cells).
- Splenic marginal zone lymphoma (± villous lymphocytes).
- Hairy cell leukemia.
- Plasmacytoma/plasma cell myeloma.
- Diffuse large B-cell lymphoma.
- Burkitt lymphoma.
T-cell and putative NK-cell neoplasms
- Precursor T-cell neoplasm: precursor T-acute lymphoblastic leukemia/LBL.
- Peripheral T-cell and NK-cell neoplasms.
- T-cell CLL/prolymphocytic leukemia.
- T-cell granular lymphocytic leukemia.
- Mycosis fungoides (including Sézary syndrome).
- Peripheral T-cell lymphoma, not otherwise characterized.
- Hepatosplenic gamma/delta T-cell lymphoma.
- Subcutaneous panniculitis-like T-cell lymphoma.
- Angioimmunoblastic T-cell lymphoma.
- Extranodal T-/NK-cell lymphoma, nasal type.
- Enteropathy-type intestinal T-cell lymphoma.
- Adult T-cell lymphoma/leukemia (human T-lymphotrophic virus [HTLV] 1+).
- Anaplastic large cell lymphoma, primary systemic type.
- Anaplastic large cell lymphoma, primary cutaneous type.
- Aggressive NK-cell leukemia.
HL
- Nodular lymphocyte-predominant HL.
- Classical HL.
- Nodular sclerosis HL.
- Lymphocyte-rich classical HL.
- Mixed-cellularity HL.
- Lymphocyte-depleted HL.
The REAL classification encompasses all the lymphoproliferative neoplasms. For more information, see the following PDQ summaries:
- Adult Acute Lymphoblastic Leukemia Treatment
- Adult Hodgkin Lymphoma Treatment
- AIDS-Related Lymphoma Treatment
- Chronic Lymphocytic Leukemia Treatment
- Hairy Cell Leukemia Treatment
- Mycosis Fungoides (Including Sézary Syndrome) Treatment
- Plasma Cell Neoplasms (Including Multiple Myeloma) Treatment
- Primary CNS Lymphoma Treatment
References:
- Zeppa P, Marino G, Troncone G, et al.: Fine-needle cytology and flow cytometry immunophenotyping and subclassification of non-Hodgkin lymphoma: a critical review of 307 cases with technical suggestions. Cancer 102 (1): 55-65, 2004.
- Young NA, Al-Saleem T: Diagnosis of lymphoma by fine-needle aspiration cytology using the revised European-American classification of lymphoid neoplasms. Cancer 87 (6): 325-45, 1999.
- National Cancer Institute sponsored study of classifications of non-Hodgkin's lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin's Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982.
- Pugh WC: Is the working formulation adequate for the classification of the low grade lymphomas? Leuk Lymphoma 10 (Suppl 1): 1-8, 1993.
- Harris NL, Jaffe ES, Stein H, et al.: A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84 (5): 1361-92, 1994.
- Pittaluga S, Bijnens L, Teodorovic I, et al.: Clinical analysis of 670 cases in two trials of the European Organization for the Research and Treatment of Cancer Lymphoma Cooperative Group subtyped according to the Revised European-American Classification of Lymphoid Neoplasms: a comparison with the Working Formulation. Blood 87 (10): 4358-67, 1996.
- Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998.
- A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997.
- Pileri SA, Milani M, Fraternali-Orcioni G, et al.: From the R.E.A.L. Classification to the upcoming WHO scheme: a step toward universal categorization of lymphoma entities? Ann Oncol 9 (6): 607-12, 1998.
- Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
Indolent NHL
Indolent non-Hodgkin lymphoma (NHL) includes the following subtypes:
- Follicular lymphoma.
- Lymphoplasmacytic lymphoma (Waldenström macroglobulinemia).
- Marginal zone lymphoma.
- Splenic marginal zone lymphoma.
- Primary cutaneous anaplastic large cell lymphoma.
Follicular Lymphoma
Follicular lymphoma comprises 20% of all NHL and as many as 70% of the indolent lymphomas reported in American and European clinical trials.[1,2,3] Most patients with follicular lymphoma are age 50 years and older and present with widespread disease at diagnosis. Nodal involvement is most common and is often accompanied by splenic and bone marrow disease. Rearrangement of the BCL2 gene is present in more than 90% of patients with follicular lymphoma; overexpression of the BCL2 protein is associated with the inability to eradicate the lymphoma by inhibiting apoptosis.[4]
Prognosis
Despite the advanced stage, the median survival ranges from 8 to 15 years, leading to the designation of being indolent.[5,6,7] Patients with advanced-stage follicular lymphoma are not cured with current therapeutic options. The rate of relapse is fairly consistent over time, even in patients who have achieved complete responses to treatment.[8] Watchful waiting, i.e., the deferring of treatment until the patient becomes symptomatic, is an option for patients with advanced-stage follicular lymphoma.[9,10] An international index for follicular lymphoma (i.e., the Follicular Lymphoma International Prognostic Index [FLIPI]) [11,12,13] identified five significant risk factors prognostic of overall survival (OS):
- Age (≤60 years vs. >60 years).
- Serum lactate dehydrogenase (LDH) (normal vs. elevated).
- Stage (stage I or stage II vs. stage III or stage IV).
- Hemoglobin level (≥120 g/L vs. <120 g/L).
- Number of nodal areas (≤4 vs. >4).
Patients with one risk factor or none have an 85% 10-year survival rate, and three or more risk factors confer a 40% 10-year survival rate.[11] In a revised FLIPI-2, an elevated beta-2-microglobulin and lymph node size of more than 6 cm are proposed prognostic factors instead of serum LDH and the number of nodal areas.[14] Although the FLIPI and FLIPI-2 indices can predict progression-free survival (PFS) and OS, the scores cannot be used to establish the need for therapy, nor can they be used to predict response to therapy.[11,14] The primary use of FLIPI or FLIPI-2 is to assure a balance of prognostic factors or to define entry requirements in randomized clinical trials. Individuals with an adverse FLIPI score may well benefit from watchful waiting or may respond well to initial therapy. An alternative prognostic index using only beta-2-microglobulin and initial bone marrow involvement (PRIMA-PI) has the disadvantage of requiring an invasive test not usually required outside the context of a clinical trial.[15] An alternative prognostic index using only noninvasive clinical variables outperformed FLIPI, FLIPI-2, and PRIMA-PI, using data from immunochemotherapy trials.[16]
Three retrospective analyses, including one pooled analysis of 5,225 patients in 13 randomized clinical trials, identified a high-risk group that had a 50% OS rate at 5 years when relapses occurred within 24 months of induction chemoimmunotherapy.[17,18,19] A fourth retrospective analysis of 296 patients who received bendamustine-rituximab found a 2-year OS rate of 38% (95% confidence interval [CI], 20%−55%) among those with progression of disease before 24 months (POD24). Most of these patients (76%) had transformed disease (histological progression to diffuse large B-cell lymphoma [DLBCL]).[20] These higher-risk POD24 patients represent a target population for clinical trials.
Follicular, small-cleaved cell lymphoma and follicular mixed small-cleaved and large cell lymphoma do not have reproducibly different disease-free survival or OS.
Therapeutic approaches
Because of the often-indolent clinical course and the lack of symptoms in some patients with follicular lymphoma, watchful waiting remains a standard of care during the initial encounter and for patients with slow asymptomatic relapsing disease. When therapy is required, numerous therapeutic options may be employed in varying sequences with an OS equivalence at 5 to 10 years.[9,21,22,23] Rituximab can be given alone or in combination with various chemotherapy options.[23,24] Rituximab can also be combined with the immunomodulating-agent lenalidomide to avoid the short- and long-term toxicities of cytotoxic agents.[25,26,27] Another anti–CD20 monoclonal antibody, obinutuzumab, can be administered with combination chemotherapy.[28] Inhibitors of phosphatidylinositol 3-kinase (PI3K) are also effective in patients with relapsed or refractory disease.[29] CD19-directed chimeric antigen receptor (CAR) T cells may be used In patients who have disease progression after two or more prior lines of therapy.[30] Mosunetuzumab, a bispecific CD20-directed CD3 T-cell engager, may also be used in this setting.[31] Consolidation therapy for relapsed disease after reinduction therapy using autologous stem cell transplant (SCT) or allogeneic SCT can be considered.[32]
Follicular lymphoma in situ and primary follicular lymphoma of the duodenum are particularly indolent variants that rarely progress and rarely require therapy.[33,34] A so-called pediatric-type nodal follicular lymphoma has indolent behavior and rarely recurs; adult patients with this histological variant are characterized by a lack of BCL2 rearrangement in conjunction with a Ki-67 proliferation index greater than 30% and a localized stage I presentation.[35]
Patients with indolent lymphoma may experience a relapse with a more aggressive histology. If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy can be performed, if feasible.[36] Documentation of conversion to a more aggressive histology requires an appropriate change to a therapy applicable to that histological type.[37] Rapid growth or discordant growth between various disease sites may indicate a histological conversion.[36] The risk of histological transformation was 30% by 10 years in a retrospective review of 325 patients from diagnosis between 1972 and 1999.[38] In this series, high-risk factors for subsequent histological transformation were advanced stage, high-risk FLIPI, and expectant management (as opposed to treatment being initiated at diagnosis). The 5-year OS rate was more than 50% for patients who had biopsy-proven, aggressive-histology transformation in several multicenter cohort studies employing rituximab plus anthracycline or platinum-based chemotherapy, or similar therapy followed by autologous or allogeneic SCT.[36,39,40]
In a prospective nonrandomized study, at a median follow-up of 6.8 years, 379 (14%) of 2,652 patients subsequently transformed to a more aggressive histology after an initial diagnosis of follicular lymphoma.[41][Level of evidence C3] The median OS after subsequent transformation was 5 years; however, among 47 patients with evidence of transformation in conjunction with follicular lymphoma at the time of initial diagnosis, the OS was no worse than that of the other nontransformed patients (5-year OS rate, 88%; 95% CI, 74%–95%).
Lymphoplasmacytic Lymphoma (Waldenström Macroglobulinemia)
Lymphoplasmacytic lymphoma is usually associated with a monoclonal serum paraprotein of immunoglobulin M (IgM) type (Waldenström macroglobulinemia).[42] Most patients have bone marrow, lymph node, and splenic involvement, and some patients may develop hyperviscosity syndrome. Most patients with Waldenström macroglobulinemia carry the MYD88 mutation, which some pathologists consider pathognomonic for the disease.[43] Other lymphomas may also be associated with serum paraproteins. Patients with lymphoplasmacytic lymphoma should be checked for associated hepatitis C virus infection.
Asymptomatic patients can be monitored for evidence of disease progression without immediate need for chemotherapy.[9,44,45]
Prognostic factors associated with symptoms requiring therapy include the following:
- Age 70 years or older.
- Beta-2-microglobulin of 3 mg/dL or more.
- Increased serum LDH.[44]
Therapeutic approaches
The management of lymphoplasmacytic lymphoma is similar to that of other low-grade lymphomas, especially diffuse, small lymphocytic lymphoma/chronic lymphocytic leukemia.[46,47,48] If the viscosity relative to water is greater than four, the patient may have manifestations of hyperviscosity. Plasmapheresis is useful for temporary, acute symptoms such as retinopathy, congestive heart failure, and central nervous system (CNS) dysfunction but can be combined with chemotherapy for prolonged disease control. Symptomatic patients with a serum viscosity of four or lower are usually started directly on chemoimmunotherapy or biologically directed therapies. Therapy may be required to correct hemolytic anemia in patients with chronic cold agglutinin disease; rituximab, bendamustine, and steroids are often used.[45] Occasionally, a heated room is required for patients whose cold agglutinins become activated by even minor chilling. Sutimlimab, an immunoglobulin G4 monoclonal antibody that selectively inhibits the complement pathway at C15, can reduce hemolysis when therapies directed at the lymphoplasmacytic lymphoma prove ineffective.[49]
First-line regimens include rituximab and ibrutinib (a Bruton tyrosine kinase [BTK] inhibitor), rituximab alone, the nucleoside analogues, and alkylating agents, either as single agents or as part of combination chemotherapy.[50,51,52,53,54] In a randomized prospective trial, 150 symptomatic patients (including previously untreated and relapsing patients) received either ibrutinib and rituximab or rituximab and a placebo. With a median follow-up of 50 months, the 4.5 year PFS rate favored the ibrutinib-and-rituximab arm (68%; 95% CI, 55%–78%) versus the rituximab-and-placebo arm (25%; 95% CI, 15%–37%) (hazard ratio, 0.25; 95% CI, 0.15–0.42; P < .0001), and the OS rate at 30 months was no different in the two arms (92%–94%).[54][Level of evidence B1] Zanubrutinib, another BTK inhibitor, was compared with ibrutinib in a randomized prospective clinical trial of 164 patients with relapsed disease and 38 previously untreated patients.[55] With a median follow-up of 18 months, the PFS rate was the same in both groups at 84%. The zanubrutinib group had fewer cases of atrial fibrillation (11 vs. 1) and 50% fewer cases of hypertension (statistics not provided).[55][Level of evidence C3] BTK inhibition with ibrutinib allowed all 13 patients with cold-antibody–mediated autoimmune hemolytic anemia and acrocyanosis to attain clinical remission regardless of underlying pathology or MYD88 status.[56][Level of evidence C3]
Previously untreated patients who received rituximab had response rates of 60% to 80%, but close monitoring of the serum IgM is required because of a sudden rise in this paraprotein at the start of therapy.[50,57,58][Level of evidence C3] The rise of IgM after rituximab can be avoided with the concomitant use of an alkylating agent, such as cyclophosphamide or the proteosome inhibitors bortezomib or ixazomib.[45,59,60,61] A combination of bortezomib, dexamethasone, and rituximab has been used with avoidance of an IgM rebound.[62,63,64] Previously untreated patients with lymphoplasmacytic lymphoma who received the nucleoside analogues cladribine and fludarabine have shown similar response rates.[53,65,66][Level of evidence C3] Patients who received single-agent alkylators, bendamustine, bortezomib, venetoclax, and combination chemotherapy with or without rituximab also show similar response rates.[53,59,61,67,68,69,70,71][Level of evidence C3] In the rare case of lymphoplasmacytic lymphoma involving the CNS (Bing-Neel syndrome), ibrutinib resulted in an 85% response rate in an anecdotal series of 28 patients.[72][Level of evidence C3]
Myeloablative therapy with autologous or allogeneic hematopoietic stem cell support is under clinical evaluation.[73,74,75,76] Candidates for this approach should avoid long-term use of alkylating agents or purine nucleoside analogues, which can deplete hematopoietic stem cells or predispose patients to myelodysplasia or acute leukemia.[50,77] After relapse from alkylating-agent therapy, 92 patients with lymphoplasmacytic lymphoma were randomly assigned to either fludarabine or cyclophosphamide, doxorubicin, and prednisone. Although relapse-free survival favored fludarabine (median duration of 19 months vs. 3 months, P < .01), no difference was observed in OS.[78][Level of evidence B1]
Marginal Zone Lymphoma
When marginal zone lymphomas involve the nodes, they are called monocytoid B-cell lymphomas or nodal marginal zone B-cell lymphomas, and when they involve extranodal sites (e.g., gastrointestinal tract, thyroid, lung, breast, orbit, and skin), they are called mucosa-associated lymphatic tissue (MALT) lymphomas.[79,80] Splenic marginal zone lymphoma is a distinct clinical entity that usually presents with massive splenomegaly. A variant form of MALT lymphoma is known as immunoproliferative small intestinal disease (IPSID).[80] A prognostic index for all of the marginal zone lymphomas has three adverse prognostic factors: age 70 years or older, stage III or stage IV disease, and high LDH level.[81] Fewer than 10% of patients transform to a higher-grade lymphoma; risk factors in one retrospective review included elevated LDH, more than four nodal sites at the time of initial diagnosis of marginal zone lymphoma, and failure to achieve complete response after initial treatment.[82]
Gastric MALT
Many patients have a history of autoimmune disease, such as Hashimoto thyroiditis or Sjögren syndrome, or of Helicobacter gastritis. Most patients present with stage I or stage II extranodal disease, which is most often in the stomach. Treatment of Helicobacter pylori infection may resolve most cases of localized gastric involvement.[83,84] After standard antibiotic regimens, 50% of patients show resolution of gastric MALT by endoscopy after 3 months. Other patients may show resolution after 12 to 18 months of observation. Of the patients who attain complete remission, 30% demonstrate monoclonality by immunoglobulin heavy chain rearrangement on stomach biopsies with a 5-year median follow-up.[85] The clinical implication of this finding is unknown. Translocation t(11;18) in patients with gastric MALT predicts for poor response to antibiotic therapy, for H. pylori –negative testing, and for poor response to oral alkylator chemotherapy.[86,87,88] Stable asymptomatic patients with persistently positive biopsies have been successfully followed on a watchful waiting approach until disease progression.[84] Patients who progress are treated with radiation therapy,[89,90,91,92,93] rituximab,[94] surgery (total gastrectomy or partial gastrectomy plus radiation therapy),[95] chemotherapy,[96] or combined-modality therapy.[97] The use of endoscopic ultrasonography may help clinicians to follow responses in these patients.[98] Four case series encompassing more than 100 patients with stage IE or IIE DLBCL with or without associated MALT (but H. pylori -positive) reported durable complete remissions in more than 50% of the patients after treatment of H. pylori.[99,100,101,102]
Extragastric MALT
Localized involvement of other sites can be treated with radiation or surgery.[90,91,92,103,104,105,106] Patients with extragastric MALT lymphoma have a higher relapse rate than patients with gastric MALT lymphoma in some series, with relapses many years and even decades later.[107] Many of these recurrences involve different MALT sites than the original location.[108] When disseminated to lymph nodes, bone marrow, or blood, this entity behaves like other low-grade lymphomas.[109,110] A prospective, randomized trial of 401 patients with nongastric, extranodal MALT compared chlorambucil alone versus rituximab plus chlorambucil versus rituximab alone.[111] With a median follow-up of 7.4 years, the event-free survival was better for the rituximab-plus-chlorambucil arm (68%) than for the rituximab-alone arm (51%) and for the chlorambucil-alone arm (50%) (P = .0009). However, the 5-year OS rate was 90% in all arms.[111] For patients with ocular adnexal MALT, antibiotic therapy using doxycycline that targeted Chlamydia psittaci resulted in durable remissions for almost half of the patients in a review of the literature that included 131 patients.[112][Level of evidence C3] These responses to doxycycline are mainly seen in Italian trials and less often in trials conducted in other geographic sites.[113] Large B-cell lymphomas of MALT sites are classified and treated as diffuse large cell lymphomas.[114] A large, retrospective review of primary ocular adnexal MALT found that after 10 years of follow-up, 4% of stage I patients treated with radiation therapy transformed to DLBCL, and 3% of them developed CNS involvement.[115]
Nodal marginal zone lymphoma
Patients with nodal marginal zone lymphoma (monocytoid B-cell lymphoma) are treated with the same paradigm of watchful waiting or therapies as described for follicular lymphoma.[116] Similar to follicular lymphoma, patients with POD24 who required initiation of therapy had a worse prognosis (53% 3-year OS rate) than did the patients without POD24 (95% 3-year OS rate).[117] Among patients with concomitant hepatitis C virus (HCV) infection, 40% to 60% attained a complete or partial remission after loss of detectable HCV RNA with antiviral treatment.[118,119][Level of evidence C3]
Mediterranean abdominal lymphoma
The disease variously known as Mediterranean abdominal lymphoma, heavy–chain disease, or IPSID, which occurs in young adults in eastern Mediterranean countries, is another version of MALT lymphoma, which responds to antibiotics in its early stages.[120]Campylobacter jejuni has been identified as one of the bacterial species associated with IPSID, and antibiotic therapy may result in remission of the disease.[121]
Splenic marginal zone lymphoma
Splenic marginal zone lymphoma is an indolent lymphoma that is marked by massive splenomegaly and peripheral blood and bone marrow involvement, usually without adenopathy.[122,123] This type of lymphoma is otherwise known as splenic lymphoma with villous lymphocytes. Splenectomy may result in prolonged remission.[124,125]
Management is similar to that of other low-grade lymphomas and usually involves rituximab alone or rituximab in combination with purine analogues or alkylating agent chemotherapy.[126] Splenic marginal zone lymphoma responds less well to chemotherapy, which would ordinarily be effective for chronic lymphocytic leukemia.[122,126,127] Among small numbers of patients with splenic marginal zone lymphoma (splenic lymphoma with villous lymphocytes) and infection with HCV, most attained a complete or partial remission after loss of detectable HCV RNA with treatment using interferon-alpha with or without ribavirin.[118,128]; [129][Level of evidence C3] In contrast, no responses to interferon were seen in six HCV-negative patients.
Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous anaplastic large cell lymphoma presents in the skin only with no preexisting lymphoproliferative disease and no extracutaneous sites of involvement.[130,131,132] Patients with this type of lymphoma encompass a spectrum ranging from clinically benign lymphomatoid papulosis, marked by localized nodules that may regress spontaneously, to a progressive and systemic disease requiring aggressive doxorubicin-based combination chemotherapy. This spectrum has been called the primary cutaneous CD30-positive T-cell lymphoproliferative disorder.
Patients with localized disease usually undergo radiation therapy. With more disseminated involvement, watchful waiting or doxorubicin-based combination chemotherapy is applied.[130,131,132]
For more information, see Chronic Lymphocytic Leukemia Treatment, Mycosis Fungoides (Including Sézary Syndrome) Treatment, Hairy Cell Leukemia Treatment, and Adult Hodgkin Lymphoma Treatment.
References:
- Armitage JO, Weisenburger DD: New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol 16 (8): 2780-95, 1998.
- A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 89 (11): 3909-18, 1997.
- Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
- López-Guillermo A, Cabanillas F, McDonnell TI, et al.: Correlation of bcl-2 rearrangement with clinical characteristics and outcome in indolent follicular lymphoma. Blood 93 (9): 3081-7, 1999.
- Peterson BA, Petroni GR, Frizzera G, et al.: Prolonged single-agent versus combination chemotherapy in indolent follicular lymphomas: a study of the cancer and leukemia group B. J Clin Oncol 21 (1): 5-15, 2003.
- Swenson WT, Wooldridge JE, Lynch CF, et al.: Improved survival of follicular lymphoma patients in the United States. J Clin Oncol 23 (22): 5019-26, 2005.
- Liu Q, Fayad L, Cabanillas F, et al.: Improvement of overall and failure-free survival in stage IV follicular lymphoma: 25 years of treatment experience at The University of Texas M.D. Anderson Cancer Center. J Clin Oncol 24 (10): 1582-9, 2006.
- Kahl BS, Yang DT: Follicular lymphoma: evolving therapeutic strategies. Blood 127 (17): 2055-63, 2016.
- Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003.
- Armitage JO, Longo DL: Is watch and wait still acceptable for patients with low-grade follicular lymphoma? Blood 127 (23): 2804-8, 2016.
- Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004.
- Perea G, Altés A, Montoto S, et al.: Prognostic indexes in follicular lymphoma: a comparison of different prognostic systems. Ann Oncol 16 (9): 1508-13, 2005.
- Buske C, Hoster E, Dreyling M, et al.: The Follicular Lymphoma International Prognostic Index (FLIPI) separates high-risk from intermediate- or low-risk patients with advanced-stage follicular lymphoma treated front-line with rituximab and the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with respect to treatment outcome. Blood 108 (5): 1504-8, 2006.
- Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009.
- Bachy E, Maurer MJ, Habermann TM, et al.: A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood 132 (1): 49-58, 2018.
- Mir F, Mattiello F, Grigg A, et al.: Follicular Lymphoma Evaluation Index (FLEX): A new clinical prognostic model that is superior to existing risk scores for predicting progression-free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol 95 (12): 1503-1510, 2020.
- Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015.
- Shi Q, Flowers CR, Hiddemann W, et al.: Thirty-Month Complete Response as a Surrogate End Point in First-Line Follicular Lymphoma Therapy: An Individual Patient-Level Analysis of Multiple Randomized Trials. J Clin Oncol 35 (5): 552-560, 2017.
- Casulo C, Dixon JG, Le-Rademacher J, et al.: Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 139 (11): 1684-1693, 2022.
- Freeman CL, Kridel R, Moccia AA, et al.: Early progression after bendamustine-rituximab is associated with high risk of transformation in advanced stage follicular lymphoma. Blood 134 (9): 761-764, 2019.
- Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d'Etude des Lymphomes Folliculaires. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1110-7, 1997.
- Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988.
- Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018.
- Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018.
- Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018.
- Leonard JP, Trnený M, Izutsu K, et al.: Augment: a phase III randomized study of lenalidomide Plus rituximab (R2) vs rituximab/placebo in patients with relapsed/refractory indolent non-Hodgkin lymphoma. [Abstract] Blood 132 (Suppl 1): A-445, 2018.
- Zucca E, Rondeau S, Vanazzi A, et al.: Short regimen of rituximab plus lenalidomide in follicular lymphoma patients in need of first-line therapy. Blood 134 (4): 353-362, 2019.
- Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017.
- Dreyling M, Santoro A, Mollica L, et al.: Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 35 (35): 3898-3905, 2017.
- Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022.
- Bartlett NL, Sehn LH, Matasar MJ, et al.: Mosunetuzumab monotherapy demonstrates durable efficacy with a manageable safety profile in patients with relapsed/refractory follicular lymphoma who received ≥2 prior therapies: updated results from a pivotal phase II study. [Abstract] Blood 140 (Suppl 1): A-610, 1467-70, 2022.
- Schaaf M, Reiser M, Borchmann P, et al.: High-dose therapy with autologous stem cell transplantation versus chemotherapy or immuno-chemotherapy for follicular lymphoma in adults. Cochrane Database Syst Rev 1: CD007678, 2012.
- Schmatz AI, Streubel B, Kretschmer-Chott E, et al.: Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol 29 (11): 1445-51, 2011.
- Jegalian AG, Eberle FC, Pack SD, et al.: Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 118 (11): 2976-84, 2011.
- Louissaint A, Ackerman AM, Dias-Santagata D, et al.: Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood 120 (12): 2395-404, 2012.
- Sarkozy C, Trneny M, Xerri L, et al.: Risk Factors and Outcomes for Patients With Follicular Lymphoma Who Had Histologic Transformation After Response to First-Line Immunochemotherapy in the PRIMA Trial. J Clin Oncol 34 (22): 2575-82, 2016.
- Tsimberidou AM, O'Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006.
- Montoto S, Davies AJ, Matthews J, et al.: Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol 25 (17): 2426-33, 2007.
- Villa D, Crump M, Panzarella T, et al.: Autologous and allogeneic stem-cell transplantation for transformed follicular lymphoma: a report of the Canadian blood and marrow transplant group. J Clin Oncol 31 (9): 1164-71, 2013.
- Williams CD, Harrison CN, Lister TA, et al.: High-dose therapy and autologous stem-cell support for chemosensitive transformed low-grade follicular non-Hodgkin's lymphoma: a case-matched study from the European Bone Marrow Transplant Registry. J Clin Oncol 19 (3): 727-35, 2001.
- Wagner-Johnston ND, Link BK, Byrtek M, et al.: Outcomes of transformed follicular lymphoma in the modern era: a report from the National LymphoCare Study (NLCS). Blood 126 (7): 851-7, 2015.
- Leblond V, Kastritis E, Advani R, et al.: Treatment recommendations from the Eighth International Workshop on Waldenström's Macroglobulinemia. Blood 128 (10): 1321-8, 2016.
- Treon SP, Xu L, Yang G, et al.: MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med 367 (9): 826-33, 2012.
- Dhodapkar MV, Hoering A, Gertz MA, et al.: Long-term survival in Waldenstrom macroglobulinemia: 10-year follow-up of Southwest Oncology Group-directed intergroup trial S9003. Blood 113 (4): 793-6, 2009.
- Ansell SM, Kyle RA, Reeder CB, et al.: Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc 85 (9): 824-33, 2010.
- Kapoor P, Ansell SM, Fonseca R, et al.: Diagnosis and Management of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) Guidelines 2016. JAMA Oncol 3 (9): 1257-1265, 2017.
- Dimopoulos MA, Kastritis E: How I treat Waldenström macroglobulinemia. Blood 134 (23): 2022-2035, 2019.
- Gertz MA: Waldenstrom Macroglobulinemia: Tailoring Therapy for the Individual. J Clin Oncol 40 (23): 2600-2608, 2022.
- Röth A, Berentsen S, Barcellini W, et al.: Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood 140 (9): 980-991, 2022.
- Gertz MA, Anagnostopoulos A, Anderson K, et al.: Treatment recommendations in Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol 30 (2): 121-6, 2003.
- Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al.: Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol 25 (22): 3344-9, 2007.
- Treon SP, Branagan AR, Ioakimidis L, et al.: Long-term outcomes to fludarabine and rituximab in Waldenström macroglobulinemia. Blood 113 (16): 3673-8, 2009.
- Leblond V, Johnson S, Chevret S, et al.: Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol 31 (3): 301-7, 2013.
- Buske C, Tedeschi A, Trotman J, et al.: Ibrutinib Plus Rituximab Versus Placebo Plus Rituximab for Waldenström's Macroglobulinemia: Final Analysis From the Randomized Phase III iNNOVATE Study. J Clin Oncol 40 (1): 52-62, 2022.
- Tam CS, Opat S, D'Sa S, et al.: A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood 136 (18): 2038-2050, 2020.
- Jalink M, Berentsen S, Castillo JJ, et al.: Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood 138 (20): 2002-2005, 2021.
- Dimopoulos MA, Zervas C, Zomas A, et al.: Treatment of Waldenström's macroglobulinemia with rituximab. J Clin Oncol 20 (9): 2327-33, 2002.
- Treon SP, Branagan AR, Hunter Z, et al.: Paradoxical increases in serum IgM and viscosity levels following rituximab in Waldenstrom's macroglobulinemia. Ann Oncol 15 (10): 1481-3, 2004.
- Dimopoulos MA, Chen C, Kastritis E, et al.: Bortezomib as a treatment option in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk 10 (2): 110-7, 2010.
- Gavriatopoulou M, García-Sanz R, Kastritis E, et al.: BDR in newly diagnosed patients with WM: final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood 129 (4): 456-459, 2017.
- Kersten MJ, Amaador K, Minnema MC, et al.: Combining Ixazomib With Subcutaneous Rituximab and Dexamethasone in Relapsed or Refractory Waldenström's Macroglobulinemia: Final Analysis of the Phase I/II HOVON124/ECWM-R2 Study. J Clin Oncol 40 (1): 40-51, 2022.
- Treon SP, Ioakimidis L, Soumerai JD, et al.: Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol 27 (23): 3830-5, 2009.
- Dimopoulos MA, García-Sanz R, Gavriatopoulou M, et al.: Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood 122 (19): 3276-82, 2013.
- Treon SP, Tripsas CK, Meid K, et al.: Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström's macroglobulinemia. Blood 124 (4): 503-10, 2014.
- Dimopoulos MA, Alexanian R: Waldenstrom's macroglobulinemia. Blood 83 (6): 1452-9, 1994.
- Laszlo D, Andreola G, Rigacci L, et al.: Rituximab and subcutaneous 2-chloro-2'-deoxyadenosine combination treatment for patients with Waldenstrom macroglobulinemia: clinical and biologic results of a phase II multicenter study. J Clin Oncol 28 (13): 2233-8, 2010.
- García-Sanz R, Montoto S, Torrequebrada A, et al.: Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol 115 (3): 575-82, 2001.
- Buske C, Hoster E, Dreyling M, et al.: The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia 23 (1): 153-61, 2009.
- Ghobrial IM, Hong F, Padmanabhan S, et al.: Phase II trial of weekly bortezomib in combination with rituximab in relapsed or relapsed and refractory Waldenstrom macroglobulinemia. J Clin Oncol 28 (8): 1422-8, 2010.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Castillo JJ, Allan JN, Siddiqi T, et al.: Venetoclax in Previously Treated Waldenström Macroglobulinemia. J Clin Oncol 40 (1): 63-71, 2022.
- Castillo JJ, Itchaki G, Paludo J, et al.: Ibrutinib for the treatment of Bing-Neel syndrome: a multicenter study. Blood 133 (4): 299-305, 2019.
- Dreger P, Glass B, Kuse R, et al.: Myeloablative radiochemotherapy followed by reinfusion of purged autologous stem cells for Waldenström's macroglobulinaemia. Br J Haematol 106 (1): 115-8, 1999.
- Desikan R, Dhodapkar M, Siegel D, et al.: High-dose therapy with autologous haemopoietic stem cell support for Waldenström's macroglobulinaemia. Br J Haematol 105 (4): 993-6, 1999.
- Martin P, Chadburn A, Christos P, et al.: Intensive treatment strategies may not provide superior outcomes in mantle cell lymphoma: overall survival exceeding 7 years with standard therapies. Ann Oncol 19 (7): 1327-30, 2008.
- Kyriakou C, Canals C, Cornelissen JJ, et al.: Allogeneic stem-cell transplantation in patients with Waldenström macroglobulinemia: report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 28 (33): 4926-34, 2010.
- Leleu X, Soumerai J, Roccaro A, et al.: Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenström macroglobulinemia treated with nucleoside analogs. J Clin Oncol 27 (2): 250-5, 2009.
- Leblond V, Lévy V, Maloisel F, et al.: Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide-doxorubicin-prednisone in 92 patients with Waldenström macroglobulinemia in first relapse or with primary refractory disease. Blood 98 (9): 2640-4, 2001.
- Zucca E, Bertoni F: The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood 127 (17): 2082-92, 2016.
- Rossi D, Bertoni F, Zucca E: Marginal-Zone Lymphomas. N Engl J Med 386 (6): 568-581, 2022.
- Thieblemont C, Cascione L, Conconi A, et al.: A MALT lymphoma prognostic index. Blood 130 (12): 1409-1417, 2017.
- Alderuccio JP, Zhao W, Desai A, et al.: Risk Factors for Transformation to Higher-Grade Lymphoma and Its Impact on Survival in a Large Cohort of Patients With Marginal Zone Lymphoma From a Single Institution. J Clin Oncol : JCO1800138, 2018.
- Zullo A, Hassan C, Andriani A, et al.: Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: a pooled data analysis. Am J Gastroenterol 104 (8): 1932-7; quiz 1938, 2009.
- Nakamura S, Sugiyama T, Matsumoto T, et al.: Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan. Gut 61 (4): 507-13, 2012.
- Wündisch T, Thiede C, Morgner A, et al.: Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 23 (31): 8018-24, 2005.
- Ye H, Liu H, Raderer M, et al.: High incidence of t(11;18)(q21;q21) in Helicobacter pylori-negative gastric MALT lymphoma. Blood 101 (7): 2547-50, 2003.
- Lévy M, Copie-Bergman C, Gameiro C, et al.: Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy. J Clin Oncol 23 (22): 5061-6, 2005.
- Nakamura S, Ye H, Bacon CM, et al.: Clinical impact of genetic aberrations in gastric MALT lymphoma: a comprehensive analysis using interphase fluorescence in situ hybridisation. Gut 56 (10): 1358-63, 2007.
- Schechter NR, Yahalom J: Low-grade MALT lymphoma of the stomach: a review of treatment options. Int J Radiat Oncol Biol Phys 46 (5): 1093-103, 2000.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Stage I and II MALT lymphoma: results of treatment with radiotherapy. Int J Radiat Oncol Biol Phys 50 (5): 1258-64, 2001.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al.: Localized mucosa-associated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 21 (22): 4157-64, 2003.
- Tsai HK, Li S, Ng AK, et al.: Role of radiation therapy in the treatment of stage I/II mucosa-associated lymphoid tissue lymphoma. Ann Oncol 18 (4): 672-8, 2007.
- De Leo AN, Bates JE, Lockney NA, et al.: Radiotherapy in Early-stage Gastric MALT: Improved Survival Without Increased Cardiac Death. Am J Clin Oncol 43 (11): 770-775, 2020.
- Martinelli G, Laszlo D, Ferreri AJ, et al.: Clinical activity of rituximab in gastric marginal zone non-Hodgkin's lymphoma resistant to or not eligible for anti-Helicobacter pylori therapy. J Clin Oncol 23 (9): 1979-83, 2005.
- Cogliatti SB, Schmid U, Schumacher U, et al.: Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 101 (5): 1159-70, 1991.
- Zinzani PL, Magagnoli M, Galieni P, et al.: Nongastrointestinal low-grade mucosa-associated lymphoid tissue lymphoma: analysis of 75 patients. J Clin Oncol 17 (4): 1254, 1999.
- Thieblemont C, Bastion Y, Berger F, et al.: Mucosa-associated lymphoid tissue gastrointestinal and nongastrointestinal lymphoma behavior: analysis of 108 patients. J Clin Oncol 15 (4): 1624-30, 1997.
- Pavlick AC, Gerdes H, Portlock CS: Endoscopic ultrasound in the evaluation of gastric small lymphocytic mucosa-associated lymphoid tumors. J Clin Oncol 15 (5): 1761-6, 1997.
- Morgner A, Miehlke S, Fischbach W, et al.: Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 19 (7): 2041-8, 2001.
- Chen LT, Lin JT, Shyu RY, et al.: Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol 19 (22): 4245-51, 2001.
- Chen LT, Lin JT, Tai JJ, et al.: Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst 97 (18): 1345-53, 2005.
- Kuo SH, Yeh KH, Wu MS, et al.: Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood 119 (21): 4838-44; quiz 5057, 2012.
- Uno T, Isobe K, Shikama N, et al.: Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: a multiinstitutional, retrospective review of 50 patients. Cancer 98 (4): 865-71, 2003.
- Bayraktar S, Bayraktar UD, Stefanovic A, et al.: Primary ocular adnexal mucosa-associated lymphoid tissue lymphoma (MALT): single institution experience in a large cohort of patients. Br J Haematol 152 (1): 72-80, 2011.
- Stefanovic A, Lossos IS: Extranodal marginal zone lymphoma of the ocular adnexa. Blood 114 (3): 501-10, 2009.
- Vazquez A, Khan MN, Sanghvi S, et al.: Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue of the salivary glands: a population-based study from 1994 to 2009. Head Neck 37 (1): 18-22, 2015.
- Raderer M, Streubel B, Woehrer S, et al.: High relapse rate in patients with MALT lymphoma warrants lifelong follow-up. Clin Cancer Res 11 (9): 3349-52, 2005.
- Sretenovic M, Colovic M, Jankovic G, et al.: More than a third of non-gastric malt lymphomas are disseminated at diagnosis: a single center survey. Eur J Haematol 82 (5): 373-80, 2009.
- Nathwani BN, Drachenberg MR, Hernandez AM, et al.: Nodal monocytoid B-cell lymphoma (nodal marginal-zone B-cell lymphoma). Semin Hematol 36 (2): 128-38, 1999.
- Raderer M, Wöhrer S, Streubel B, et al.: Assessment of disease dissemination in gastric compared with extragastric mucosa-associated lymphoid tissue lymphoma using extensive staging: a single-center experience. J Clin Oncol 24 (19): 3136-41, 2006.
- Zucca E, Conconi A, Martinelli G, et al.: Final Results of the IELSG-19 Randomized Trial of Mucosa-Associated Lymphoid Tissue Lymphoma: Improved Event-Free and Progression-Free Survival With Rituximab Plus Chlorambucil Versus Either Chlorambucil or Rituximab Monotherapy. J Clin Oncol 35 (17): 1905-1912, 2017.
- Kiesewetter B, Raderer M: Antibiotic therapy in nongastrointestinal MALT lymphoma: a review of the literature. Blood 122 (8): 1350-7, 2013.
- Grünberger B, Hauff W, Lukas J, et al.: 'Blind' antibiotic treatment targeting Chlamydia is not effective in patients with MALT lymphoma of the ocular adnexa. Ann Oncol 17 (3): 484-7, 2006.
- Kuo SH, Chen LT, Yeh KH, et al.: Nuclear expression of BCL10 or nuclear factor kappa B predicts Helicobacter pylori-independent status of early-stage, high-grade gastric mucosa-associated lymphoid tissue lymphomas. J Clin Oncol 22 (17): 3491-7, 2004.
- Desai A, Joag MG, Lekakis L, et al.: Long-term course of patients with primary ocular adnexal MALT lymphoma: a large single-institution cohort study. Blood 129 (3): 324-332, 2017.
- Thieblemont C, Molina T, Davi F: Optimizing therapy for nodal marginal zone lymphoma. Blood 127 (17): 2064-71, 2016.
- Luminari S, Merli M, Rattotti S, et al.: Early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL-NF10 study. Blood 134 (10): 798-801, 2019.
- Vallisa D, Bernuzzi P, Arcaini L, et al.: Role of anti-hepatitis C virus (HCV) treatment in HCV-related, low-grade, B-cell, non-Hodgkin's lymphoma: a multicenter Italian experience. J Clin Oncol 23 (3): 468-73, 2005.
- Merli M, Rattotti S, Spina M, et al.: Direct-Acting Antivirals as Primary Treatment for Hepatitis C Virus-Associated Indolent Non-Hodgkin Lymphomas: The BArT Study of the Fondazione Italiana Linfomi. J Clin Oncol 40 (35): 4060-4070, 2022.
- Isaacson PG: Gastrointestinal lymphoma. Hum Pathol 25 (10): 1020-9, 1994.
- Lecuit M, Abachin E, Martin A, et al.: Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med 350 (3): 239-48, 2004.
- Arcaini L, Paulli M, Boveri E, et al.: Splenic and nodal marginal zone lymphomas are indolent disorders at high hepatitis C virus seroprevalence with distinct presenting features but similar morphologic and phenotypic profiles. Cancer 100 (1): 107-15, 2004.
- Arcaini L, Rossi D, Paulli M: Splenic marginal zone lymphoma: from genetics to management. Blood 127 (17): 2072-81, 2016.
- Bertoni F, Zucca E: State-of-the-art therapeutics: marginal-zone lymphoma. J Clin Oncol 23 (26): 6415-20, 2005.
- Parry-Jones N, Matutes E, Gruszka-Westwood AM, et al.: Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patients. Br J Haematol 120 (5): 759-64, 2003.
- Arcaini L, Lazzarino M, Colombo N, et al.: Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood 107 (12): 4643-9, 2006.
- Iannitto E, Ambrosetti A, Ammatuna E, et al.: Splenic marginal zone lymphoma with or without villous lymphocytes. Hematologic findings and outcomes in a series of 57 patients. Cancer 101 (9): 2050-7, 2004.
- Hermine O, Lefrère F, Bronowicki JP, et al.: Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N Engl J Med 347 (2): 89-94, 2002.
- Kelaidi C, Rollot F, Park S, et al.: Response to antiviral treatment in hepatitis C virus-associated marginal zone lymphomas. Leukemia 18 (10): 1711-6, 2004.
- de Bruin PC, Beljaards RC, van Heerde P, et al.: Differences in clinical behaviour and immunophenotype between primary cutaneous and primary nodal anaplastic large cell lymphoma of T-cell or null cell phenotype. Histopathology 23 (2): 127-35, 1993.
- Willemze R, Beljaards RC: Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders. A proposal for classification and guidelines for management and treatment. J Am Acad Dermatol 28 (6): 973-80, 1993.
- Kempf W, Pfaltz K, Vermeer MH, et al.: EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 118 (15): 4024-35, 2011.
Aggressive NHL
Aggressive non-Hodgkin lymphoma (NHL) includes the following subtypes:
- Diffuse large B-cell lymphoma.
- Mediastinal large B-cell lymphoma (primary mediastinal large B-cell lymphoma).
- Follicular large cell lymphoma.
- Anaplastic large cell lymphoma.
- Extranodal NK–/T-cell lymphoma.
- Lymphomatoid granulomatosis.
- Angioimmunoblastic T-cell lymphoma.
- Peripheral T-cell lymphoma.
- Enteropathy-type intestinal T-cell lymphoma.
- Intravascular large B-cell lymphoma (intravascular lymphomatosis).
- Burkitt lymphoma/diffuse small noncleaved-cell lymphoma.
- Lymphoblastic lymphoma.
- Adult T-cell leukemia/lymphoma.
- Mantle cell lymphoma.
- Polymorphic posttransplantation lymphoproliferative disorder.
- True histiocytic lymphoma.
- Primary effusion lymphoma.
- Plasmablastic lymphoma.
Diffuse Large B-cell Lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common type of NHL and comprises 30% of newly diagnosed cases.[1] Most patients present with rapidly enlarging masses, often with both local and systemic symptoms (designated B symptoms with fever, recurrent night sweats, or weight loss). For more information about weight loss, see Nutrition in Cancer Care.
Some cases of large B-cell lymphoma have a prominent background of reactive T cells and often of histiocytes, so-called T-cell/histiocyte-rich large B-cell lymphoma. This subtype of large cell lymphoma has frequent liver, spleen, and bone marrow involvement; however, the outcome is equivalent to that of similarly staged patients with DLBCL.[2,3,4] Some patients with DLBCL at diagnosis have a concomitant indolent small B-cell component; while overall survival (OS) appears similar after multidrug chemotherapy, there is a higher risk of indolent relapse.[5]
Prognosis
Most patients with localized disease are curable with combined-modality therapy or combination chemotherapy alone.[6] Among patients with advanced-stage disease, 50% are cured with doxorubicin-based combination chemotherapy and rituximab, typically R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).[7,8,9]
The National Comprehensive Cancer Network International Prognostic Index (IPI) for aggressive NHL (diffuse large cell lymphoma) identifies the following five significant risk factors prognostic of OS and their associated risk scores:[10]
- Age.
- <40 years: 0.
- 41–60 years: 1.
- 61–75 years: 2.
- >75 years: 3.
- Stage III/IV: 1.
- Performance status 2/3/4: 1.
- Serum lactate dehydrogenase (LDH).
- Normalized: 0.
- >1x–3x: 1.
- >3x: 2.
- Number of extranodal sites ≥2: 1.
Risk scores:
- Low (0 or 1): 5-year OS rate, 96%; progression-free survival (PFS) rate, 91%.
- Low intermediate (2 or 3): 5-year OS rate, 82%; PFS rate, 74%.
- High intermediate (4 or 5): 5-year OS rate, 64%; PFS rate, 51%.
- High (>6): 5-year OS rate, 33%; PFS rate, 30%.
Age-adjusted and stage-adjusted modifications of this IPI are used for younger patients with localized disease.[11] Shorter intervals of time between diagnosis and treatment appear to be a surrogate for poor prognostic biological factors.[12]
The BCL2 gene and rearrangement of the MYC gene or dual overexpression of the MYC gene, or both, confer a particularly poor prognosis.[13,14,15] Dose-intensive therapies, infusional therapies, and stem cell transplantation (SCT) consolidation are being explored in this high-risk group.[16,17] A retrospective review evaluated 159 patients with previously untreated DLBCL who underwent double-hit genetic testing by fluorescence in situ hybridization (FISH) and achieved complete response (CR).[18] The induction therapy did not alter 3-year relapse-free survival or OS when autologous SCT was employed.
In a retrospective review of 117 patients with relapsed or refractory DLBCL who underwent autologous SCT, the 4-year OS rate was 25% for double-hit lymphomas (rearrangement of BCL2 and MYC), 61% for double-expressor lymphomas (no rearrangement, but increased expression of BCL2 and MYC), and 70% for patients without these features.[19] Patients at high risk of relapse may be considered for clinical trials.[20]
Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.[21] For example, true anaplastic lymphoma kinase (ALK)-positive large B-cell lymphomas are extremely rare, and they do not respond well to conventional R-CHOP therapy. Anecdotal responses to ALK inhibitors like lorlatinib or alectinib have been reported.[22][Level of evidence C3] Patients who have DLBCL with coexpression of CD20 and CD30 may define a subgroup with a unique molecular signature, a more favorable prognosis, and possible therapeutic implication for the use of anti-CD30–specific therapy, such as brentuximab vedotin.[23] Patients with DLBCL who are event-free after 2 years have a subsequent OS equivalent to that of the age- and sex-matched general population.[24]
Central nervous system (CNS) prophylaxis
The CNS-IPI tool predicts which patients have a CNS relapse risk exceeding 10%. It was developed by the German Lymphoma Study Group and validated by the British Columbia Cancer Agency database. The presence of four to six of the CNS-IPI risk factors (age >60 years, performance status ≥2, elevated LDH, stage III or IV disease, >1 extranodal site, or involvement of the kidneys or adrenal glands) was used to define a high-risk group for CNS recurrence (a 12% risk of CNS involvement by 2 years).[25]
CNS prophylaxis (usually with four to six doses of intrathecal methotrexate) is often recommended for patients with testicular involvement.[26,27,28][Level of evidence C3] A retrospective analysis of the German RICOVER studies compared intrathecal methotrexate with no prophylaxis in patients with DLBCL. This study was completed during the R-CHOP treatment era. With the possible exception of patients with testicular involvement, the analysis showed that intrathecal methotrexate did not reduce the risk of CNS disease.[29][Level of evidence C3] Some clinicians employ high-dose intravenous (IV) methotrexate (usually four doses) as an alternative to intrathecal therapy because drug delivery is improved and patient morbidity is decreased.[30] A retrospective study evaluated 1,162 patients from 21 U.S. academic centers where 77% received intrathecal methotrexate, 20% received high-dose IV methotrexate, and 3% received both sequentially (because of toxicity).[31] There was no difference in CNS relapse rates between patients who received intrathecal methotrexate or high-dose IV methotrexate (5.4% vs. 6.8%, P = .40). Testicular involvement, nongerminal center subtype, and high extranodal involvement predicted increased CNS relapse regardless of the route of prophylaxis.[31] Two retrospective studies evaluating high-dose methotrexate in patients with high-risk DLBCL also showed no improvement in CNS relapse rate.[32,33][Level of evidence C3] Patients deemed at high risk for CNS relapse (e.g., patients with four to six CNS-IPI risk factors) often receive intrathecal methotrexate or high-dose IV methotrexate, but the lack of confirmatory randomized studies calls this standard into question and shows an urgent need for better therapeutics verified in clinical trials. Patients with testicular involvement are an exception, as they show benefit from intrathecal or high-dose IV methotrexate.[26,27,28][Level of evidence C3]
The addition of rituximab to cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP)-based regimens has significantly reduced the risk of CNS relapse in retrospective analyses.[29,34][Level of evidence C3] Patients with CNS dissemination at diagnosis or at relapse usually receive rituximab and high doses of methotrexate and/or cytarabine followed by autologous SCT, but this approach has not been assessed in randomized trials.[35,36][Level of evidence C3]
Primary Mediastinal Large B-cell Lymphoma
Primary mediastinal (thymic) large B-cell lymphoma (PMBCL) is a subset of DLBCL with molecular characteristics that are most similar to nodular-sclerosing Hodgkin lymphoma (HL).[37] Mediastinal lymphomas with features intermediate between primary mediastinal B-cell lymphoma and nodular-sclerosing HL are called mediastinal gray-zone lymphomas.[38,39] Patients are usually female and young (median age, 30–40 years). Patients present with a locally invasive anterior mediastinal mass that may cause respiratory symptoms or superior vena cava syndrome.
Prognosis and therapy are the same as for other comparably staged patients with DLBCL. Uncontrolled, phase II studies employing dose-adjusted R-EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin plus rituximab) or R-CHOP show high cure rates while avoiding any mediastinal radiation.[39,40,41,42,43,44,45][Level of evidence C1] These results suggest that patients who receive R-CHOP–based regimens may avoid the serious long-term complications of radiation therapy when given with chemotherapy. Posttreatment fluorine F 18-fludeoxyglucose (18F-FDG) positron emission tomography–computed tomography (PET-CT) scans are controversial; it remains unclear if PET scans can reliably identify patients who can take or omit radiation therapy consolidation.[40,46,47,48]
A retrospective review of 109 patients with PMBCL showed that 63% had a negative end-of-treatment PET-CT (EOT-PET-CT) (Deauville score 1–3).[49] No radiation therapy was offered and the 5-year time-to-progression rate (similar to disease-free survival, but restricted to lymphoma relapse) was 90%, and the 5-year OS rate was 97%.[49][Level of evidence C3] Patients with a positive EOT-PET-CT received radiation therapy consolidation. It is unclear from this study whether those patients might have done just as well without radiation therapy. Clinicians may follow improvement over time for Deauville 4 scores on EOT-PET-CT scans as an alternative to radiation therapy, but this has not been studied in a clinical trial.
In situations where mediastinal radiation therapy would encompass the left side of the heart or would increase breast cancer risk in young female patients, proton therapy may be considered to reduce radiation dose to organs at risk.[50] For more information, see the Superior Vena Cava Syndrome section in Cardiopulmonary Syndromes.
Because PMBCL is characterized by high expression of programmed death-ligand 1 (PD-L1) and variable expression of CD30, a phase II study evaluated nivolumab plus brentuximab vedotin in 30 patients with relapsed disease. With a median follow-up of 11.1 months, the objective response rate was 73% (95% CI, 54%−88%).[51][Level of evidence C3] Similarly, phase I and II trials of pembrolizumab in 74 patients with relapsed or refractory disease showed an objective response rate of 45% to 48%. The median duration of response was not reached for the 21 patients with a median follow-up of 29 months or for the 53 patients with a median follow-up of 12.5 months.[52][Level of evidence C3]
Among those who had received two prior lines of therapy, more than one-half of patients who received CAR T-cell therapy with lisocabtagene maraleucel had disease response.[53][Level of evidence C3]
Follicular Large Cell Lymphoma
Prognosis
The natural history of follicular large cell lymphoma remains controversial.[54] While there is agreement about the significant number of long-term disease-free survivors with early-stage disease, the curability of patients with advanced disease (stage III or stage IV) remains uncertain. Some groups report a continuous relapse rate similar to the other follicular lymphomas (a pattern of indolent lymphoma).[55] Other investigators report a plateau in freedom from progression at levels expected for an aggressive lymphoma (40% at 10 years).[56,57] This discrepancy may be caused by variations in histological classification between institutions and the rarity of patients with follicular large cell lymphoma. A retrospective review of 252 patients, all treated with anthracycline-containing combination chemotherapy, showed that patients with more than 50% diffuse components on biopsy had a worse OS than other patients with follicular large cell lymphoma.[58]
Therapeutic approaches
Treatment of follicular large cell lymphoma is more similar to treatment of aggressive NHL than it is to the treatment of indolent NHL. In support of this approach, treatment with high-dose chemotherapy and autologous hematopoietic peripheral SCT shows the same curative potential in patients with follicular large cell lymphoma who relapse as it does in patients with diffuse large cell lymphoma who relapse.[59][Level of evidence C1]
Among those who had received two prior lines of therapy, more than one-half of patients who received CAR T-cell therapy with lisocabtagene maraleucel had disease response.[53][Level of evidence C3]
Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphoma (ALCL) is a T-cell lymphoma associated with the CD30 antigen. The translocation of chromosomes 2 and 5 creates a unique fusion protein with a nucleophosmin-ALK.[60,61] Patients whose lymphomas express ALK by immunohistochemistry are usually younger and may have systemic symptoms, extranodal disease, and advanced-stage disease. However, they have a more favorable survival rate than ALK-negative patients.[62,63]
A prospective randomized trial included 452 patients with CD30-positive T-cell lymphoma, 70% of whom had ALCL (22% ALK-positive and 48% ALK-negative patients). The trial compared the previously used standard regimen, CHOP, with brentuximab vedotin (an anti-CD30 monoclonal antibody conjugated to a cytotoxic agent) combined with cyclophosphamide, doxorubicin, and prednisone.[64] With a median follow-up of 35 months, the 3-year OS rate was 77% in the brentuximab vedotin arm and 68% in the CHOP arm (hazard ratio [HR], 0.66; 95% CI, 0.46–0.95; P = .02).[64][Level of evidence A1] This established brentuximab vedotin plus cyclophosphamide, doxorubicin, and prednisone as a new option for patients with ALCL and other CD30-positive T-cell lymphomas, such as angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, not otherwise specified. For patients with relapsed disease, anecdotal responses have been reported for brentuximab vedotin,[65,66,67,68] romidepsin,[69] and pralatrexate.[70][Level of evidence C3]
In a phase II study (NCT00866047), 66% of 58 patients attained a complete response with brentuximab vedotin. At a median follow-up of 58 months, the 5-year PFS rate was 57% (95% CI, 41%–74%), and the 5-year OS rate was 79% (95% CI, 65%–92%). Of the patients achieving a complete response, 42% underwent hematopoietic SCT.[68][Level of evidence C3] In a retrospective review, 39 patients with relapsed disease had a 3-year PFS rate of 50% after autologous or allogeneic SCT.[71][Level of evidence C2] A retrospective review of 84 patients with ALK-negative ALCL suggested a survival benefit with autologous SCT. This hypothesis requires confirmation in a randomized prospective trial.[72]
ALCL in children is usually characterized by systemic and cutaneous disease and has high response rates and good OS with doxorubicin-based combination chemotherapy.[73] Patients with breast implant–associated ALCL may do well without chemotherapy after capsulectomy and implant removal if the disease is confined to the fibrous capsule, and no associated mass or lymphadenopathy is present.[74,75,76,77] Most patients with breast implant–associated ALCL have a characteristic deletion at 20Q13.13 that may help diagnostically to distinguish it from cutaneous or systemic ALCL.[78]
Primary cutaneous ALCL is a distinct entity that is typically ALK-negative and has a very indolent/low-grade clinical course.
Extranodal Natural Killer (NK)-/T-cell Lymphoma
Extranodal natural killer (NK)-/T-cell lymphoma (nasal type) is an aggressive lymphoma marked by extensive necrosis and angioinvasion, most often presenting in extranodal sites, in particular the nasal or paranasal sinus region.[79] Other extranodal sites include the palate, trachea, skin, and gastrointestinal tract. Hemophagocytic syndrome may occur; historically, these tumors were considered part of lethal midline granuloma.[80] In most cases, Epstein-Barr virus (EBV) genomes are detectable in the tumor cells and immunophenotyping shows CD56 positivity. Cases with blood and marrow involvement are considered NK-cell leukemia.
The increased risk of CNS involvement and of local recurrence has led to recommendations for radiation therapy locally, concurrently, before the start of chemotherapy or between cycle two and three of chemotherapy, and for intrathecal prophylaxis and/or prophylactic cranial radiation therapy.[81,82,83,84,85,86,87,88]
A retrospective review of 1,273 early-stage patients stratified them into a low-risk group and high-risk group using stage, age, LDH, performance status, and primary tumor invasion. Low-risk patients fared best with radiation therapy alone,[89] while high-risk patients fared best with a strategy of radiation therapy combined with chemotherapy.[87,90,91]
In a retrospective review of 303 previously untreated patients from an international consortium who received nonanthracycline chemotherapy, the OS rates were identical for early-stage patients (72%−74% at 5 years) who received either concurrent chemotherapy and radiation therapy or chemotherapy followed by radiation therapy.[92][Level of evidence C3]
Higher doses of radiation therapy administered at more than 50 Gy are associated with improved outcomes according to anecdotal reports.[87] The highly aggressive course, with poor response and short survival with standard therapies, especially for patients with advanced-stage disease or extranasal presentation, has led some investigators to recommend autologous or allogeneic peripheral SCT consolidation.[88,93,94,95,96,97] Asparaginase-containing regimens have shown anecdotal response rates greater than 50% for relapsing, refractory, or newly diagnosed patients.[88,98,99,100,101][Level of evidence C3] Because of the lack of randomized clinical trials with more than 100 patients for this rare type of T-cell lymphoma, regimens containing pegaspargase have become the standard for systemic therapy. Pegaspargase is a less toxic formulation of asparaginase with less hypersensitivity reactions and a longer half-life.[102,103] NK-/T-cell lymphoma that presents only in the skin has a more favorable prognosis, especially in patients with coexpression of CD30 with CD56.[104] A benign NK-cell enteropathy (EBV negative) on endoscopic biopsy can be distinguished from NK-/T-cell lymphoma.[105] In a phase II trial, the anti-PD-L1 antibody avelumab was given to 21 patients with relapsed or refractory disease. The CR rate was 24%, the overall response rate was 38%, and responses correlated with tumor PD-L1 expression.[106][Level of evidence C3] Treatment with pembrolizumab, an anti-programmed cell death protein 1 (PD-1) antibody, resulted in similar responses in patients with relapsed or refractory disease.[107][Level of evidence C3]
Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis is an EBV-positive large B-cell lymphoma with a predominant T-cell background.[108,109] The histology shows association with angioinvasion and vasculitis, usually manifesting as pulmonary lesions or paranasal sinus involvement.
Patients are managed like others with diffuse large cell lymphoma and require doxorubicin-based combination chemotherapy.
Angioimmunoblastic T-cell Lymphoma
Angioimmunoblastic T-cell lymphoma (AITL or ATCL) was formerly called angioimmunoblastic lymphadenopathy with dysproteinemia. Characterized by clonal T-cell receptor gene rearrangement, this entity is managed like diffuse large cell lymphoma.[110,111,112,113] Patients present with profound lymphadenopathy, fever, night sweats, weight loss, skin rash, a positive Coombs test, and polyclonal hypergammaglobulinemia.[80] Opportunistic infections are frequent because of an underlying immune deficiency. B-cell EBV genomes are detected in most affected patients.[114] For more information about weight loss, see Nutrition in Cancer Care and for more information about skin rash, see Pruritus.
Doxorubicin-based combination chemotherapy, such as the CHOP regimen, is recommended as it is for other aggressive lymphomas.[110,113] For CD30-positive cases, brentuximab combined with cyclophosphamide, doxorubicin, and prednisone is the standard of care.[64][Level of evidence B1] For more information, see the Anaplastic Large Cell Lymphoma section. The International Peripheral T-Cell Lymphoma Project involving 22 international centers identified 243 patients with AITL or ATCL; the 5-year OS and failure-free survival rates were 33% and 18%, respectively.[115] Myeloablative chemotherapy and radiation therapy with autologous or allogeneic peripheral stem cell support has been described in anecdotal reports.[72,96,116,117,118] Anecdotal responses have been reported for cyclosporine,[119] pralatrexate,[120] bendamustine,[121] the histone deacetylase inhibitor romidepsin, and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[69,122][Level of evidence C3] Occasional spontaneous remissions and protracted responses to steroids only have been reported.
Peripheral T-cell Lymphoma
Patients with peripheral T-cell lymphoma have diffuse large cell or diffuse mixed lymphoma that expresses a cell surface phenotype of a postthymic (or peripheral) T-cell expressing CD4 or CD8 but not both together.[123] Peripheral T-cell lymphoma encompasses a group of heterogeneous nodal T-cell lymphomas that will require future delineation.[80,124] This includes the so-called Lennert lymphoma, a T-cell lymphoma admixed with a preponderance of lymphoepithelioid cells.
Prognosis
Most investigators report worse response and survival rates for patients with peripheral T-cell lymphomas than for patients with comparably staged B-cell aggressive lymphomas.[124,125] Most patients present with multiple adverse prognostic factors (i.e., older age, stage IV, multiple extranodal sites, and elevated LDH), and these patients have a low (<20%) failure-free survival and OS at 5 years.[124,125] As with other lymphomas (e.g., DLBCL or follicular lymphoma), event-free survival at 24 months predicts a 5-year OS of 78%.[126]
Therapeutic approaches
Therapy involves doxorubicin-based combination chemotherapy (such as CHOP or CHOPE [CHOP plus etoposide]), which is also used for DLBCL.[127] For CD30-positive cases, brentuximab combined with cyclophosphamide, doxorubicin, and prednisone is the standard of care.[64][Level of evidence B1] For more information, see the Anaplastic Large Cell Lymphoma section. For patients with early-stage disease, anecdotal retrospective series disagree on the value of consolidative radiation therapy after combination chemotherapy.[128][Level of evidence C3] Consolidation therapy using high-dose chemotherapy with autologous or allogeneic hematopoietic stem cell support has been given to patients with advanced-stage peripheral T-cell lymphoma after induction therapy in multiple phase II or retrospective trials. Evidence for this approach is anecdotal.[72,96,116,118,129,130,131][Level of evidence C3]
A randomized prospective trial included 104 patients younger than 61 years with stage II, III, or IV peripheral T-cell lymphoma (excluding ALK-positive ALCL). Patients received either autologous SCT or allogeneic SCT as consolidation therapy after induction with CHOEP (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone) followed by DHAP (dexamethasone, cytarabine, and cisplatin).[132][Level of evidence C3] With a median follow-up of 42 months, the 3-year EFS rate was 43% for patients who received allogeneic SCT and 38% for patients who received autologous SCT. The 3-year OS rate was 57% for patients who received allogeneic SCT and 70% for patients who received autologous SCT (P = nonsignificant). None of the 21 responding patients who proceeded to allogeneic SCT relapsed, and 36% of patients who proceeded to autologous SCT relapsed. Eight of 26 patients (31%) who received allogeneic SCT died of graft-versus-host disease, and none of the 41 patients who received autologous SCT died of toxicity. The benefit of graft-versus-lymphoma effect was negated by increased transplant-related mortality.
In a prospective trial of 109 evaluable patients with relapsing disease, treatment with pralatrexate resulted in a 30% response rate and a median 10-month duration of response.[69,133][Level of evidence C3] Similar response rates were seen in 130 evaluable patients with relapsing disease who received romidepsin in a prospective trial.[69][Level of evidence C3] Anecdotal responses have been seen with a combination of pralatrexate and romidepsin,[120] single-agent bendamustine,[121] belinostat,[134] and brentuximab vedotin (even if there is little or no CD30 expression on the lymphoma).[122][Level of evidence C3] Incorporation of these new agents with CHOP chemotherapy is under clinical evaluation.[64,124]
An unusual type of peripheral T-cell lymphoma occurring mostly in young men, hepatosplenic T-cell lymphoma, appears to be localized to the hepatic and splenic sinusoids, with cell surface expression of the T-cell receptor gamma/delta.[135,136,137] Another variant, subcutaneous panniculitis-like T-cell lymphoma, is localized to subcutaneous tissue associated with hemophagocytic syndrome.[138,139,140,141] These patients have cells that express alpha-beta phenotype. Those with gamma-delta phenotype have a more aggressive clinical course and are classified as cutaneous gamma-delta T-cell lymphoma.[142,143,144] These patients may manifest involvement of the epidermis, dermis, subcutaneous region, or mucosa. These entities have extremely poor prognoses with an extremely aggressive clinical course and are treated within the same paradigm as the highest-risk groups with DLBCL.[96,137] An indolent T-cell lymphoproliferative disease of the gastrointestinal tract must be distinguished from peripheral T-cell lymphoma because no therapy may be indicated.[145]
Enteropathy-type Intestinal T-cell Lymphoma
Enteropathy-type intestinal T-cell lymphoma involves the small bowel of patients with gluten-sensitive enteropathy (celiac sprue).[80,146,147,148] Because a gluten-free diet prevents the development of lymphoma, patients diagnosed with celiac sprue in childhood rarely develop lymphoma. The diagnosis of celiac disease is usually made by finding villous atrophy in the resected intestine. Surgery is often required for diagnosis and to avoid perforation during therapy.
Therapy is with doxorubicin-based combination chemotherapy, but relapse rates appear higher than for comparably staged diffuse large cell lymphoma.[147,148,149] Complications of treatment include gastrointestinal bleeding, small bowel perforation, and enterocolic fistulae; patients often require parenteral nutrition. For more information on parenteral nutrition, see Nutrition in Cancer Care. Multifocal intestinal perforations and visceral abdominal involvement are seen at the time of relapse. High-dose therapy with hematopoietic stem cell rescue has been applied in first remission or at relapse.[96,147,150][Level of evidence C2] Evidence for this approach is anecdotal.
Intravascular Large B-cell Lymphoma (Intravascular Lymphomatosis)
Intravascular lymphomatosis is characterized by large cell lymphoma confined to the intravascular lumen. The brain, kidneys, lungs, and skin are the organs most likely affected by intravascular lymphomatosis.
With the use of aggressive R-CHOP–based combination chemotherapy, as is used in DLBCL, the prognosis is similar to that of conventional stage IV DLBCL.[151,152,153]
Burkitt Lymphoma/Diffuse Small Noncleaved-cell Lymphoma
Burkitt lymphoma/diffuse small noncleaved-cell lymphoma typically involves younger patients and represents the most common type of pediatric NHL.[154] These types of aggressive extranodal B-cell lymphomas are characterized by translocation and deregulation of the MYC gene on chromosome 8.[155] A subgroup of patients with dual translocation of MYC and BCL2 appear to have an extremely poor outcome despite aggressive therapy (median OS, 5 months).[156][Level of evidence C1]
In some patients with larger B cells, there is morphological overlap with DLBCL. These Burkitt-like large cell lymphomas show MYC deregulation, extremely high proliferation rates, and a gene-expression profile as expected for classic Burkitt lymphoma.[157,158,159] Endemic cases, usually from Africa, involve the facial bones or jaws of children, mostly containing EBV genomes. Sporadic cases usually involve the gastrointestinal system, ovaries, or kidneys. Patients present with rapidly growing masses and a very high LDH but are potentially curable with intensive doxorubicin-based combination chemotherapy.
Therapeutic approaches
Treatment of Burkitt lymphoma/diffuse small noncleaved-cell lymphoma involves aggressive multidrug regimens in combination with rituximab, similar to those used for the advanced-stage aggressive lymphomas (diffuse large cell).[160,161,162,163] Aggressive combination chemotherapy, which is patterned after that used in childhood Burkitt lymphoma, has been very successful for adult patients with more than 60% of advanced-stage patients free of disease at 5 years.[164,165,166,167] Adverse prognostic factors include bulky abdominal disease and high serum LDH. Patients with Burkitt lymphoma have a 20% to 30% lifetime risk of CNS involvement. Prophylaxis with intrathecal chemotherapy is required as part of induction therapy.[168] Patients with HIV-associated Burkitt lymphoma also benefit from less-toxic modification of the aggressive multidrug regimens in combination with rituximab.[169][Level of evidence C3] For more information, see Primary CNS Lymphoma Treatment and AIDS-Related Lymphoma Treatment.
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (precursor T-cell) is a very aggressive form of NHL. It often, but not exclusively, occurs in young patients.[170] It is commonly associated with large mediastinal masses and has a high predilection for disseminating to bone marrow and the CNS.
Treatment is usually patterned after that for acute lymphoblastic leukemia. Intensive combination chemotherapy with or without bone marrow transplantation is the standard treatment for this aggressive histological type of NHL.[171,172,173] Radiation therapy is sometimes given to areas of bulky tumor masses. Because these forms of NHL tend to progress quickly, combination chemotherapy is instituted rapidly once the diagnosis has been confirmed. Careful review of the pathological specimens, bone marrow aspirate, biopsy specimen, cerebrospinal fluid cytology, and lymphocyte marker constitute the most important aspects of the pretreatment staging workup. For more information, see Adult Acute Lymphoblastic Leukemia Treatment.
Adult T-cell Leukemia/Lymphoma
Adult T-cell leukemia/lymphoma (ATL) is caused by infection with the retrovirus human T-lymphotrophic virus 1 and is frequently associated with lymphadenopathy, hypercalcemia, circulating leukemic cells, bone and skin involvement, hepatosplenomegaly, a rapidly progressive course, and poor response to combination chemotherapy.[174,175] ATL has been divided into four clinical subtypes:[176,177]
- Acute (aggressive course with leukemia, with or without extranodal or nodal involvement).
- Lymphoma (aggressive course with lymphadenopathy and no leukemia).
- Chronic (indolent course with leukemia and lymphadenopathy).
- Smoldering (indolent course with only leukemia).
The acute and lymphoma types of ATL have done poorly with strategies of combination chemotherapy and allogeneic SCT with a median OS under 1 year.[178,179,180] Using combination chemotherapy, less than 10% of 807 patients were alive after 4 years.[180] Anecdotal durable remissions have been reported after allogeneic SCT and even after subsequent donor lymphocyte infusion for relapses after transplant.[181][Level of evidence C3] Among 815 patients who underwent allogeneic SCT in two retrospective reviews, the 3-year OS rates were 36% and 26%.[182,183][Level of evidence C1]
The combination of zidovudine and interferon-alpha has activity against ATL, even for patients who failed previous cytotoxic therapy. Durable remissions are seen in most patients who present with this combination, but are not seen in patients with the lymphoma subtype of ATL.[184,185,186,187,188] In a multicenter phase II study of 26 relapsed patients, 42% responded to lenalidomide (including four CR).[189][Level of evidence C3] Symptomatic local progression of all subtypes responds well to palliative radiation therapy.[190] In the relapsed setting, an overall response rate above 50% was seen using mogamulizumab, a humanized monoclonal antibody against the C-C chemokine receptor 4 (CCR4).[191][Level of evidence C3] For CD30-positive cases, brentuximab combined with cyclophosphamide, doxorubicin, and prednisone is the standard of care.[64][Level of evidence B1] For more information, see the Anaplastic Large Cell Lymphoma section.
Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is found in lymph nodes, the spleen, bone marrow, blood, and sometimes the gastrointestinal system (lymphomatous polyposis).[192] MCL is characterized by CD5-positive follicular mantle B cells, a translocation of chromosomes 11 and 14, and an overexpression of the cyclin D1 protein.[192] MCL may be divided into two clinical subtypes: a classical version with lymphadenopathy with high SOX-11 expression that manifests with an aggressive clinical course and a worse prognosis versus a leukemic, non-nodal version with low SOX-11 expression and a more indolent course and a better prognosis.[193] A complex karyotype predicts poor response to induction therapy and inferior survival.[194] There is frequent overlap on presentation with these subtypes, and the therapeutic implication remains unclear. However, both of these versions can converge later in their course into a blastoid phenotype or treatment-resistant phenotype due to genomic instability and selection.[195,196]
Like the low-grade lymphomas, MCL appears incurable with anthracycline-based chemotherapy and occurs in older patients with generally asymptomatic advanced-stage disease. The median survival, however, is significantly shorter (5–7 years) than that of other lymphomas, and this histology is now considered to be an aggressive lymphoma.[197] A diffuse pattern and the blastoid variant have an aggressive course with shorter survival, while the mantle zone type may have a more indolent course.[196,198] A high cell-proliferation rate (increased Ki-67, mitotic index, beta-2-microglobulin) may be associated with a poorer prognosis.[199,200]
Therapeutic approaches
Asymptomatic patients with low-risk scores on the IPI may do well when initial therapy is deferred.[201,202][Level of evidence C3] There is no standard approach to MCL. Several induction chemotherapy regimens may be employed for symptomatic progressing disease. These regimens range in intensity from rituximab alone to rituximab plus ibrutinib, rituximab plus bendamustine, R-CHOP, or high-dose intensive regimens such as R-hyper C-VAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with methotrexate and cytarabine). Some physicians use autologous SCT or allogeneic SCT consolidation next, while others prefer rituximab maintenance, reserving high-dose consolidation for a later time.[203] Ibrutinib, lenalidomide, and bortezomib have shown activity in relapsing patients, and these drugs are being incorporated up front.[204,205,206,207,208,209]
It is unclear which therapeutic approach offers the best long-term survival in this clinicopathological entity.
In a phase II trial of previously untreated patients with MCL older than 64 years, 50 patients received the B-cell receptor-inhibitor ibrutinib plus rituximab. With a median follow-up of 45 months, the overall response rate was 96%, the CR rate was 76%, the 3-year PFS rate was 87%, and the 3-year OS rate was 94%.[209][Level of evidence C3] In a phase II trial of 131 previously untreated patients with MCL aged 65 years or younger, 1 year of ibrutinib plus 4 weeks of rituximab resulted in a CR rate of 89% prior to any chemotherapy consolidation.[210][Level of evidence C3] Another phase II trial using ibrutinib plus rituximab included asymptomatic patients with previously untreated MCL; the CR rate was 87%.[211][Level of evidence C3] Previously treated patients who received ibrutinib had a response rate of 86% (21% CR rate) and a median PFS of 14 months.[206][Level of evidence C3] In a prospective randomized trial, 280 patients with relapsed or refractory MCL received either ibrutinib or temsirolimus.[212] With a median follow-up of 15 months, the median PFS favored ibrutinib (14.6 months vs. 6.2 months; HR, 0.43; 95% CI, 0.32–0.58, P < .0001).[212][Level of evidence B1] Ibrutinib was combined with another active agent, venetoclax, in a phase II study of 23 patients with relapsed or refractory MCL.[213] An unprecedented 71% of patients had a CR and 78% of responding patients maintained response at 15 months.[213][Level of evidence C3]
A prospective randomized trial included 523 patients aged 65 years and older with MCL. Patients were randomly assigned to receive either ibrutinib, bendamustine, and rituximab or bendamustine and rituximab alone.[214] With a median follow-up of 84.7 months, the median PFS was 80.6 months for patients who received ibrutinib, and 52.9 months for patients who received bendamustine and rituximab alone (HR, 0.75; 95% CI, 0.59–0.96; P = .01). There was no difference in the 7-year OS rate (55.0% vs. 56.8%; HR, 1.07; 95% CI, 0.81–1.40).[214][Level of evidence B1] It is unclear if patients who received ibrutinib alone could have achieved these same results without receiving conventional chemotherapy. The magnitude of benefit demonstrated by the PFS results contrasted with the insufficient OS benefit after 7 years may cast doubt on the long-term safety of this combination.
In a prospective randomized trial, 560 patients older than 60 years and not eligible for SCT were given either R-CHOP or R-FC (rituximab, fludarabine, cyclophosphamide) for six to eight cycles, followed by maintenance therapy in responders randomly assigned to rituximab or interferon-alpha maintenance therapy.[215] With a median follow-up of 7.6 years, the median OS was significantly shorter after R-FC than after R-CHOP (3.9 years versus 6.4 years; P = .0054).[215][Level of evidence A1] In the same trial, with a median follow-up of 8 years for the 316 responding patients, rituximab maintenance resulted in improved OS over interferon maintenance (median OS, 9.8 years vs. 7.1 years; P = .009).[215][Level of evidence A1] Patients responsive to R-CHOP benefitted most from rituximab in OS (median 9.8 years vs. 6.4 years; P = .0026).[215][Level of evidence C1] A randomized trial compared bendamustine plus rituximab (BR) with R-CHOP and showed improved PFS (35 vs. 22 months; HR, 0.49; 95% CI, 0.28–0.79; P = .004) but no difference in OS.[216][Level of evidence B1] However, this trial failed to show any benefit for rituximab maintenance after BR. A prospective randomized trial of 487 patients compared VR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin, prednisone) with R-CHOP.[217] With a median follow-up of 82 months, the median OS was longer for VR-CAP (90.7 months) compared with R-CHOP (55.7 months) (HR, 0.66; 95% CI, 0.51−0.85; P = .001).[217][Level of evidence A1]
A prospective randomized trial of 497 patients younger than 65 years compared six cycles of R-CHOP with six cycles of alternating R-CHOP and R-DHAP (rituximab, dexamethasone, cytarabine, and cisplatin), with both groups then receiving autologous SCT.[218,219][Level of evidence B1] With a median follow-up of 10.6 years, the 10-year PFS rate was 73% for patients who received R-DHAP and 57% for patients who received R-CHOP (HR, 0.56; P = .038), but there was no difference in the 10-year OS rates (60% [R-DHAP] vs. 55% [R-CHOP]; HR, 0.80; 0.61–1.06; P = .12).[220][Level of evidence B1] This is the randomized trial referenced by all subsequent articles establishing a role for cytarabine in induction therapy; the ultimate lack of survival advantage casts doubt on this assertion.
Randomized trials have not confirmed an OS benefit in patients who receive consolidation therapy with autologous or allogeneic SCT.[221,222,223,224,225,226]
In a prospective trial (NCT00921414) of 299 patients who were previously untreated for MCL, 257 responders received four courses of R-DHAP and autologous SCT. The patients were randomly assigned to receive rituximab maintenance therapy for 3 years versus no maintenance therapy. After randomization, a median follow-up at 50.2 months showed the rate of PFS at 4-years favored the rituximab-maintenance arm at 83% (95% CI, 73%–88%) versus the no-maintenance arm at 64% (95% CI, 55%–73%; P < .001). The 4-year OS rate also favored the rituximab-maintenance arm at 89% (95% CI, 81%–94%) versus the no-maintenance arm at 80% (95% CI, 72%–88%; P = .04).[227][Level of evidence A1]
Lenalidomide with or without rituximab also shows response rates of around 50% in relapsed patients, with even higher response rates for previously untreated patients.[205,208,228,229][Level of evidence C3]
Acalabrutinib (another B-cell receptor inhibitor via the Bruton tyrosine kinase [BTK] pathway) was studied in 124 patients with relapsed or refractory MCL.[230] In a phase II study, there was an 81% overall response rate, 40% CR rate, and 67% 1-year PFS rate.[230][Level of evidence C3] Similarly, the BTK inhibitor zanubrutinib was evaluated in a phase II study of 86 patients with relapsed or refractory MCL.[231] After a median follow-up of 35.3 months, the overall response rate was 84%, the CR rate was 78%, and the median PFS was 33.0 months.[231][Level of evidence C3]
Patients with relapsed or refractory MCL whose disease did not respond to ibrutinib or acalabrutinib were enrolled in a phase II trial using brexucabtagene autoleucel, an anti-CD19 CAR T-cell therapy.[232] With a median follow-up of 36 months, 74 patients had an objective response rate of 91% (95% CI, 82%−97%) and a CR rate of 68% (95% CI, 55%−78%).[232][Level of evidence C3] Grade 3 or higher cytokine release syndrome occurred in 15% of patients, and neurological events occurred in 31% of patients.
In summary, the optimal sequencing of these various therapies is unclear and is the subject of an ongoing Intergroup clinical trial. Rituximab, lenalidomide, ibrutinib, acalabrutinib, venetoclax, and zanubrutinib represent directed biological agents that may lead to chemotherapy-free treatment strategies for patients with MCL.[233]
Routine administration of CNS prophylaxis in high-risk MCL has never been studied in a prospective randomized trial. The use of intrathecal or high-dose methotrexate or the use of systemic therapies with CNS penetration like ibrutinib, high-dose cytarabine, or venetoclax, have not been studied and proven efficacious in this situation.[196]
Posttransplantation Lymphoproliferative Disorder
Patients who undergo transplantation of the heart, lung, liver, kidney, or pancreas usually require lifelong immunosuppression. This may result in posttransplantation lymphoproliferative disorder (PTLD) in 1% to 3% of recipients, which appears as an aggressive lymphoma.[234] Pathologists can distinguish a polyclonal B-cell hyperplasia from a monoclonal B-cell lymphoma; both are almost always associated with EBV.[235]
Prognosis
Poor performance status, grafted organ involvement, high IPI, elevated LDH, and multiple sites of disease are poor prognostic factors for PTLD.[236,237]
Therapeutic options
In some cases, withdrawal of immunosuppression results in eradication of the lymphoma.[238,239] When this is unsuccessful or not feasible, a course of rituximab may be considered, because it has shown durable remissions in approximately 60% of patients and a favorable toxicity profile.[238,240,241] If these measures fail, doxorubicin-based combination chemotherapy (R-CHOP) is recommended, although some patients can avoid cytotoxic therapy.[241,242] Localized presentations can be controlled with surgery or radiation therapy alone. These localized mass lesions, which may grow over a period of months, are often phenotypically polyclonal and tend to occur within weeks or a few months after transplantation.[235] Multifocal, rapidly progressive disease occurs late after transplantation (>1 year) and is usually phenotypically monoclonal and associated with EBV.[243] These patients may have durable remissions using standard chemotherapy regimens for aggressive lymphoma.[243,244,245] Instances of EBV-negative PTLD occur even later (median, 5 years posttransplant) and have a worse prognosis; R-CHOP chemotherapy can be applied directly in this circumstance.[246] A sustained clinical response after failure from chemotherapy was attained using an immunotoxin (anti-CD22 B-cell surface antigen antibody linked with ricin, a plant toxin).[247] An anti-interleukin-6 monoclonal antibody is also under clinical evaluation.[248]
True Histiocytic Lymphoma
True histiocytic lymphomas are very rare tumors that show histiocytic differentiation and express histiocytic markers in the absence of B-cell or T-cell lineage-specific immunologic markers.[249,250] Care must be taken with immunophenotypic tests to exclude ALCL or hemophagocytic syndromes caused by viral infections, especially EBV.
Therapeutic options
Therapy is modeled after the treatment of comparably staged diffuse large cell lymphomas, but the optimal approach remains to be defined.
Primary Effusion Lymphoma
Primary effusion lymphoma presents exclusively or mainly in the pleural, pericardial, or abdominal cavities in the absence of an identifiable tumor mass.[251] Patients are usually HIV seropositive, and the tumor usually contains Kaposi sarcoma–associated herpes virus/human herpes virus 8.[252]
Prognosis
The prognosis of primary effusion lymphoma is extremely poor.
Therapeutic approaches
Therapy is usually modeled after the treatment of comparably staged diffuse large cell lymphomas.
Plasmablastic Lymphoma
Plasmablastic lymphoma is most often seen in patients with HIV infection and is characterized by CD20-negative large B cells with plasmacytic features. This type of lymphoma has a very aggressive clinical course, including poor responses and short remissions with standard chemotherapy.[253] Anecdotal reports suggest using aggressive chemotherapy for Burkitt or lymphoblastic lymphoma, followed by SCT consolidation in responding patients, when feasible.[253,254,255]
References:
- Sehn LH, Salles G: Diffuse Large B-Cell Lymphoma. N Engl J Med 384 (9): 842-858, 2021.
- Delabie J, Vandenberghe E, Kennes C, et al.: Histiocyte-rich B-cell lymphoma. A distinct clinicopathologic entity possibly related to lymphocyte predominant Hodgkin's disease, paragranuloma subtype. Am J Surg Pathol 16 (1): 37-48, 1992.
- Achten R, Verhoef G, Vanuytsel L, et al.: T-cell/histiocyte-rich large B-cell lymphoma: a distinct clinicopathologic entity. J Clin Oncol 20 (5): 1269-77, 2002.
- Bouabdallah R, Mounier N, Guettier C, et al.: T-cell/histiocyte-rich large B-cell lymphomas and classical diffuse large B-cell lymphomas have similar outcome after chemotherapy: a matched-control analysis. J Clin Oncol 21 (7): 1271-7, 2003.
- Ghesquières H, Berger F, Felman P, et al.: Clinicopathologic characteristics and outcome of diffuse large B-cell lymphomas presenting with an associated low-grade component at diagnosis. J Clin Oncol 24 (33): 5234-41, 2006.
- Miller TP, Dahlberg S, Cassady JR, et al.: Chemotherapy alone compared with chemotherapy plus radiotherapy for localized intermediate- and high-grade non-Hodgkin's lymphoma. N Engl J Med 339 (1): 21-6, 1998.
- Coiffier B, Lepage E, Briere J, et al.: CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346 (4): 235-42, 2002.
- Coiffier B: State-of-the-art therapeutics: diffuse large B-cell lymphoma. J Clin Oncol 23 (26): 6387-93, 2005.
- Habermann TM, Weller EA, Morrison VA, et al.: Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 24 (19): 3121-7, 2006.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Scott DW, King RL, Staiger AM, et al.: High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131 (18): 2060-2064, 2018.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- Staiger AM, Ziepert M, Horn H, et al.: Clinical Impact of the Cell-of-Origin Classification and the MYC/ BCL2 Dual Expresser Status in Diffuse Large B-Cell Lymphoma Treated Within Prospective Clinical Trials of the German High-Grade Non-Hodgkin's Lymphoma Study Group. J Clin Oncol 35 (22): 2515-2526, 2017.
- Howlett C, Snedecor SJ, Landsburg DJ, et al.: Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 170 (4): 504-14, 2015.
- Sesques P, Johnson NA: Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood 129 (3): 280-288, 2017.
- Landsburg DJ, Falkiewicz MK, Maly J, et al.: Outcomes of Patients With Double-Hit Lymphoma Who Achieve First Complete Remission. J Clin Oncol 35 (20): 2260-2267, 2017.
- Herrera AF, Mei M, Low L, et al.: Relapsed or Refractory Double-Expressor and Double-Hit Lymphomas Have Inferior Progression-Free Survival After Autologous Stem-Cell Transplantation. J Clin Oncol 35 (1): 24-31, 2017.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
- Soumerai JD, Rosenthal A, Harkins S, et al.: Next-generation ALK inhibitors are highly active in ALK-positive large B-cell lymphoma. Blood 140 (16): 1822-1826, 2022.
- Hu S, Xu-Monette ZY, Balasubramanyam A, et al.: CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood 121 (14): 2715-24, 2013.
- Maurer MJ, Ghesquières H, Jais JP, et al.: Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol 32 (10): 1066-73, 2014.
- Schmitz N, Zeynalova S, Nickelsen M, et al.: CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 34 (26): 3150-6, 2016.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Boehme V, Schmitz N, Zeynalova S, et al.: CNS events in elderly patients with aggressive lymphoma treated with modern chemotherapy (CHOP-14) with or without rituximab: an analysis of patients treated in the RICOVER-60 trial of the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL). Blood 113 (17): 3896-902, 2009.
- Glantz MJ, Cole BF, Recht L, et al.: High-dose intravenous methotrexate for patients with nonleukemic leptomeningeal cancer: is intrathecal chemotherapy necessary? J Clin Oncol 16 (4): 1561-7, 1998.
- Orellana-Noia VM, Reed DR, McCook AA, et al.: Single-route CNS prophylaxis for aggressive non-Hodgkin lymphomas: real-world outcomes from 21 US academic institutions. Blood 139 (3): 413-423, 2022.
- Puckrin R, El Darsa H, Ghosh S, et al.: Ineffectiveness of high-dose methotrexate for prevention of CNS relapse in diffuse large B-cell lymphoma. Am J Hematol 96 (7): 764-771, 2021.
- Jeong H, Cho H, Kim H, et al.: Efficacy and safety of prophylactic high-dose MTX in high-risk DLBCL: a treatment intent-based analysis. Blood Adv 5 (8): 2142-2152, 2021.
- Villa D, Connors JM, Shenkier TN, et al.: Incidence and risk factors for central nervous system relapse in patients with diffuse large B-cell lymphoma: the impact of the addition of rituximab to CHOP chemotherapy. Ann Oncol 21 (5): 1046-52, 2010.
- Ferreri AJ, Donadoni G, Cabras MG, et al.: High Doses of Antimetabolites Followed by High-Dose Sequential Chemoimmunotherapy and Autologous Stem-Cell Transplantation in Patients With Systemic B-Cell Lymphoma and Secondary CNS Involvement: Final Results of a Multicenter Phase II Trial. J Clin Oncol 33 (33): 3903-10, 2015.
- Schmitz N, Wu HS: Advances in the Treatment of Secondary CNS Lymphoma. J Clin Oncol 33 (33): 3851-3, 2015.
- Savage KJ: Primary mediastinal large B-cell lymphoma. Blood 140 (9): 955-970, 2022.
- van Besien K, Kelta M, Bahaguna P: Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 19 (6): 1855-64, 2001.
- Dunleavy K, Wilson WH: Primary mediastinal B-cell lymphoma and mediastinal gray zone lymphoma: do they require a unique therapeutic approach? Blood 125 (1): 33-9, 2015.
- Dunleavy K, Pittaluga S, Maeda LS, et al.: Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 368 (15): 1408-16, 2013.
- Savage KJ, Yenson PR, Shenkier T, et al.: The outcome of primary mediastinal large B-cell lymphoma in the R-CHOP treatment era. [Abstract] Blood 120 (21): A-303, 2012.
- Vassilakopoulos TP, Pangalis GA, Katsigiannis A, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone with or without radiotherapy in primary mediastinal large B-cell lymphoma: the emerging standard of care. Oncologist 17 (2): 239-49, 2012.
- Rieger M, Osterborg A, Pettengell R, et al.: Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol 22 (3): 664-70, 2011.
- Gleeson M, Hawkes EA, Cunningham D, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone (R-CHOP) in the management of primary mediastinal B-cell lymphoma: a subgroup analysis of the UK NCRI R-CHOP 14 versus 21 trial. Br J Haematol 175 (4): 668-672, 2016.
- Held G, Thurner L, Poeschel V, et al.: Role of radiotherapy and dose-densification of R-CHOP in primary mediastinal B-cell lymphoma: A subgroup analysis of the unfolder trial of the German Lymphoma Alliance (GLA). [Abstract] J Clin Oncol 38 (Suppl 15): A-8041, 2021.
- Martelli M, Ceriani L, Zucca E, et al.: [18F]fluorodeoxyglucose positron emission tomography predicts survival after chemoimmunotherapy for primary mediastinal large B-cell lymphoma: results of the International Extranodal Lymphoma Study Group IELSG-26 Study. J Clin Oncol 32 (17): 1769-75, 2014.
- Zinzani PL, Broccoli A, Casadei B, et al.: The role of rituximab and positron emission tomography in the treatment of primary mediastinal large B-cell lymphoma: experience on 74 patients. Hematol Oncol 33 (4): 145-50, 2015.
- Ceriani L, Martelli M, Conconi A, et al.: Prognostic models for primary mediastinal (thymic) B-cell lymphoma derived from 18-FDG PET/CT quantitative parameters in the International Extranodal Lymphoma Study Group (IELSG) 26 study. Br J Haematol 178 (4): 588-591, 2017.
- Hayden AR, Tonseth P, Lee DG, et al.: Outcome of primary mediastinal large B-cell lymphoma using R-CHOP: impact of a PET-adapted approach. Blood 136 (24): 2803-2811, 2020.
- Dabaja BS, Hoppe BS, Plastaras JP, et al.: Proton therapy for adults with mediastinal lymphomas: the International Lymphoma Radiation Oncology Group guidelines. Blood 132 (16): 1635-1646, 2018.
- Zinzani PL, Santoro A, Gritti G, et al.: Nivolumab Combined With Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety From the Phase II CheckMate 436 Study. J Clin Oncol 37 (33): 3081-3089, 2019.
- Armand P, Rodig S, Melnichenko V, et al.: Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J Clin Oncol 37 (34): 3291-3299, 2019.
- Abramson JS, Palomba ML, Gordon LI, et al.: Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396 (10254): 839-852, 2020.
- Longo DL: What's the deal with follicular lymphomas? J Clin Oncol 11 (2): 202-8, 1993.
- Anderson JR, Vose JM, Bierman PJ, et al.: Clinical features and prognosis of follicular large-cell lymphoma: a report from the Nebraska Lymphoma Study Group. J Clin Oncol 11 (2): 218-24, 1993.
- Bartlett NL, Rizeq M, Dorfman RF, et al.: Follicular large-cell lymphoma: intermediate or low grade? J Clin Oncol 12 (7): 1349-57, 1994.
- Wendum D, Sebban C, Gaulard P, et al.: Follicular large-cell lymphoma treated with intensive chemotherapy: an analysis of 89 cases included in the LNH87 trial and comparison with the outcome of diffuse large B-cell lymphoma. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (4): 1654-63, 1997.
- Hans CP, Weisenburger DD, Vose JM, et al.: A significant diffuse component predicts for inferior survival in grade 3 follicular lymphoma, but cytologic subtypes do not predict survival. Blood 101 (6): 2363-7, 2003.
- Vose JM, Bierman PJ, Lynch JC, et al.: Effect of follicularity on autologous transplantation for large-cell non-Hodgkin's lymphoma. J Clin Oncol 16 (3): 844-9, 1998.
- Bai RY, Ouyang T, Miething C, et al.: Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 96 (13): 4319-27, 2000.
- Hapgood G, Savage KJ: The biology and management of systemic anaplastic large cell lymphoma. Blood 126 (1): 17-25, 2015.
- Gascoyne RD, Aoun P, Wu D, et al.: Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 93 (11): 3913-21, 1999.
- Sibon D, Fournier M, Brière J, et al.: Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol 30 (32): 3939-46, 2012.
- Horwitz S, O'Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019.
- Younes A, Bartlett NL, Leonard JP, et al.: Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363 (19): 1812-21, 2010.
- Pro B, Advani R, Brice P, et al.: Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30 (18): 2190-6, 2012.
- Prince HM, Kim YH, Horwitz SM, et al.: Brentuximab vedotin or physician's choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 390 (10094): 555-566, 2017.
- Pro B, Advani R, Brice P, et al.: Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 130 (25): 2709-2717, 2017.
- Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012.
- O'Connor OA, Horwitz S, Hamlin P, et al.: Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 27 (26): 4357-64, 2009.
- Smith SM, Burns LJ, van Besien K, et al.: Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol 31 (25): 3100-9, 2013.
- Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022.
- Seidemann K, Tiemann M, Schrappe M, et al.: Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma: a report of the Berlin-Frankfurt-Münster Group Trial NHL-BFM 90. Blood 97 (12): 3699-706, 2001.
- Miranda RN, Aladily TN, Prince HM, et al.: Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol 32 (2): 114-20, 2014.
- Clemens MW, Medeiros LJ, Butler CE, et al.: Complete Surgical Excision Is Essential for the Management of Patients With Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 34 (2): 160-8, 2016.
- Mehta-Shah N, Clemens MW, Horwitz SM: How I treat breast implant-associated anaplastic large cell lymphoma. Blood 132 (18): 1889-1898, 2018.
- Jaffe ES, Ashar BS, Clemens MW, et al.: Best Practices Guideline for the Pathologic Diagnosis of Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 38 (10): 1102-1111, 2020.
- Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020.
- Tse E, Kwong YL: How I treat NK/T-cell lymphomas. Blood 121 (25): 4997-5005, 2013.
- Rizvi MA, Evens AM, Tallman MS, et al.: T-cell non-Hodgkin lymphoma. Blood 107 (4): 1255-64, 2006.
- Li YX, Yao B, Jin J, et al.: Radiotherapy as primary treatment for stage IE and IIE nasal natural killer/T-cell lymphoma. J Clin Oncol 24 (1): 181-9, 2006.
- Lee J, Suh C, Park YH, et al.: Extranodal natural killer T-cell lymphoma, nasal-type: a prognostic model from a retrospective multicenter study. J Clin Oncol 24 (4): 612-8, 2006.
- Li CC, Tien HF, Tang JL, et al.: Treatment outcome and pattern of failure in 77 patients with sinonasal natural killer/T-cell or T-cell lymphoma. Cancer 100 (2): 366-75, 2004.
- Yamaguchi M, Tobinai K, Oguchi M, et al.: Phase I/II study of concurrent chemoradiotherapy for localized nasal natural killer/T-cell lymphoma: Japan Clinical Oncology Group Study JCOG0211. J Clin Oncol 27 (33): 5594-600, 2009.
- Kim SJ, Kim K, Kim BS, et al.: Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-Cell Lymphoma: Consortium for Improving Survival of Lymphoma study. J Clin Oncol 27 (35): 6027-32, 2009.
- Li YX, Fang H, Liu QF, et al.: Clinical features and treatment outcome of nasal-type NK/T-cell lymphoma of Waldeyer ring. Blood 112 (8): 3057-64, 2008.
- Vargo JA, Patel A, Glaser SM, et al.: The impact of the omission or inadequate dosing of radiotherapy in extranodal natural killer T-cell lymphoma, nasal type, in the United States. Cancer 123 (16): 3176-3185, 2017.
- Yamaguchi M, Suzuki R, Oguchi M: Advances in the treatment of extranodal NK/T-cell lymphoma, nasal type. Blood 131 (23): 2528-2540, 2018.
- Yang Y, Cao JZ, Lan SM, et al.: Association of Improved Locoregional Control With Prolonged Survival in Early-Stage Extranodal Nasal-Type Natural Killer/T-Cell Lymphoma. JAMA Oncol 3 (1): 83-91, 2017.
- Yang Y, Zhu Y, Cao JZ, et al.: Risk-adapted therapy for early-stage extranodal nasal-type NK/T-cell lymphoma: analysis from a multicenter study. Blood 126 (12): 1424-32; quiz 1517, 2015.
- Yamaguchi M, Suzuki R, Oguchi M, et al.: Treatments and Outcomes of Patients With Extranodal Natural Killer/T-Cell Lymphoma Diagnosed Between 2000 and 2013: A Cooperative Study in Japan. J Clin Oncol 35 (1): 32-39, 2017.
- Kwong YL, Kim SJ, Tse E, et al.: Sequential chemotherapy/radiotherapy was comparable with concurrent chemoradiotherapy for stage I/II NK/T-cell lymphoma. Ann Oncol 29 (1): 256-263, 2018.
- Liang R, Todd D, Chan TK, et al.: Treatment outcome and prognostic factors for primary nasal lymphoma. J Clin Oncol 13 (3): 666-70, 1995.
- Cheung MM, Chan JK, Lau WH, et al.: Primary non-Hodgkin's lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16 (1): 70-7, 1998.
- Hausdorff J, Davis E, Long G, et al.: Non-Hodgkin's lymphoma of the paranasal sinuses: clinical and pathological features, and response to combined-modality therapy. Cancer J Sci Am 3 (5): 303-11, 1997 Sep-Oct.
- Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
- Au WY, Weisenburger DD, Intragumtornchai T, et al.: Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood 113 (17): 3931-7, 2009.
- Jaccard A, Gachard N, Marin B, et al.: Efficacy of L-asparaginase with methotrexate and dexamethasone (AspaMetDex regimen) in patients with refractory or relapsing extranodal NK/T-cell lymphoma, a phase 2 study. Blood 117 (6): 1834-9, 2011.
- Yamaguchi M, Kwong YL, Kim WS, et al.: Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol 29 (33): 4410-6, 2011.
- Li JW, Li YJ, Zhong MZ, et al.: Efficacy and tolerance of GELOXD/P-GEMOXD in newly diagnosed nasal-type extranodal NK/T-cell lymphoma: A multicenter retrospective study. Eur J Haematol 100 (3): 247-256, 2018.
- Wei L, Wang L, Cong J, et al.: SVILE regimen, a combination of dexamethasone, vindesine, ifosfamide, pegaspargase, and etoposide, for treating relapsed/refractory extranodal natural killer/T-cell lymphoma, nasal type. Leuk Res 96: 106422, 2020.
- Liang R, Gao GX, Chen JP, et al.: A phase 2 study of methotrexate, etoposide, dexamethasone, and pegaspargase chemotherapy for newly diagnosed, relapsed, or refractory extranodal natural killer/T-cell lymphoma, nasal type: a multicenter trial in Northwest China. Hematol Oncol 35 (4): 619-629, 2017.
- Wang X, Zhang L, Liu X, et al.: Efficacy and Safety of a Pegasparaginase-Based Chemotherapy Regimen vs an L-asparaginase-Based Chemotherapy Regimen for Newly Diagnosed Advanced Extranodal Natural Killer/T-Cell Lymphoma: A Randomized Clinical Trial. JAMA Oncol 8 (7): 1035-1041, 2022.
- Mraz-Gernhard S, Natkunam Y, Hoppe RT, et al.: Natural killer/natural killer-like T-cell lymphoma, CD56+, presenting in the skin: an increasingly recognized entity with an aggressive course. J Clin Oncol 19 (8): 2179-88, 2001.
- Mansoor A, Pittaluga S, Beck PL, et al.: NK-cell enteropathy: a benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: clinicopathologic features and follow-up in a unique case series. Blood 117 (5): 1447-52, 2011.
- Kim SJ, Lim JQ, Laurensia Y, et al.: Avelumab for the treatment of relapsed or refractory extranodal NK/T-cell lymphoma: an open-label phase 2 study. Blood 136 (24): 2754-2763, 2020.
- Kwong YL, Chan TSY, Tan D, et al.: PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 129 (17): 2437-2442, 2017.
- Guinee D, Jaffe E, Kingma D, et al.: Pulmonary lymphomatoid granulomatosis. Evidence for a proliferation of Epstein-Barr virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am J Surg Pathol 18 (8): 753-64, 1994.
- Myers JL, Kurtin PJ, Katzenstein AL, et al.: Lymphomatoid granulomatosis. Evidence of immunophenotypic diversity and relationship to Epstein-Barr virus infection. Am J Surg Pathol 19 (11): 1300-12, 1995.
- Siegert W, Agthe A, Griesser H, et al.: Treatment of angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma using prednisone with or without the COPBLAM/IMVP-16 regimen. A multicenter study. Kiel Lymphoma Study Group. Ann Intern Med 117 (5): 364-70, 1992.
- Jaffe ES: Angioimmunoblastic T-cell lymphoma: new insights, but the clinical challenge remains. Ann Oncol 6 (7): 631-2, 1995.
- Siegert W, Nerl C, Agthe A, et al.: Angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol 6 (7): 659-64, 1995.
- Lunning MA, Vose JM: Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood 129 (9): 1095-1102, 2017.
- Bräuninger A, Spieker T, Willenbrock K, et al.: Survival and clonal expansion of mutating "forbidden" (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic t cell lymphoma. J Exp Med 194 (7): 927-40, 2001.
- Federico M, Rudiger T, Bellei M, et al.: Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol 31 (2): 240-6, 2013.
- Reimer P, Rüdiger T, Geissinger E, et al.: Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 27 (1): 106-13, 2009.
- Kyriakou C, Canals C, Finke J, et al.: Allogeneic stem cell transplantation is able to induce long-term remissions in angioimmunoblastic T-cell lymphoma: a retrospective study from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 27 (24): 3951-8, 2009.
- Park SI, Horwitz SM, Foss FM, et al.: The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer 125 (9): 1507-1517, 2019.
- Advani R, Horwitz S, Zelenetz A, et al.: Angioimmunoblastic T cell lymphoma: treatment experience with cyclosporine. Leuk Lymphoma 48 (3): 521-5, 2007.
- Amengual JE, Lichtenstein R, Lue J, et al.: A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131 (4): 397-407, 2018.
- Damaj G, Gressin R, Bouabdallah K, et al.: Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol 31 (1): 104-10, 2013.
- Fanale MA, Horwitz SM, Forero-Torres A, et al.: Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 131 (19): 2120-2124, 2018.
- Rüdiger T, Weisenburger DD, Anderson JR, et al.: Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin's Lymphoma Classification Project. Ann Oncol 13 (1): 140-9, 2002.
- Weisenburger DD, Savage KJ, Harris NL, et al.: Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the International Peripheral T-cell Lymphoma Project. Blood 117 (12): 3402-8, 2011.
- Sonnen R, Schmidt WP, Müller-Hermelink HK, et al.: The International Prognostic Index determines the outcome of patients with nodal mature T-cell lymphomas. Br J Haematol 129 (3): 366-72, 2005.
- Maurer MJ, Ellin F, Srour L, et al.: International Assessment of Event-Free Survival at 24 Months and Subsequent Survival in Peripheral T-Cell Lymphoma. J Clin Oncol 35 (36): 4019-4026, 2017.
- Carson KR, Horwitz SM, Pinter-Brown LC, et al.: A prospective cohort study of patients with peripheral T-cell lymphoma in the United States. Cancer 123 (7): 1174-1183, 2017.
- Briski R, Feldman AL, Bailey NG, et al.: Survival in patients with limited-stage peripheral T-cell lymphomas. Leuk Lymphoma 56 (6): 1665-70, 2015.
- Rodriguez J, Munsell M, Yazji S, et al.: Impact of high-dose chemotherapy on peripheral T-cell lymphomas. J Clin Oncol 19 (17): 3766-70, 2001.
- d'Amore F, Relander T, Lauritzsen GF, et al.: Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol 30 (25): 3093-9, 2012.
- Schmitz N, Lenz G, Stelljes M: Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood 132 (3): 245-253, 2018.
- Schmitz N, Truemper L, Bouabdallah K, et al.: A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood 137 (19): 2646-2656, 2021.
- O'Connor OA, Pro B, Pinter-Brown L, et al.: Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29 (9): 1182-9, 2011.
- O'Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015.
- Belhadj K, Reyes F, Farcet JP, et al.: Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood 102 (13): 4261-9, 2003.
- Chanan-Khan A, Islam T, Alam A, et al.: Long-term survival with allogeneic stem cell transplant and donor lymphocyte infusion following salvage therapy with anti-CD52 monoclonal antibody (Campath) in a patient with alpha/beta hepatosplenic T-cell non-Hodgkin's lymphoma. Leuk Lymphoma 45 (8): 1673-5, 2004.
- Pro B, Allen P, Behdad A: Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood 136 (18): 2018-2026, 2020.
- Go RS, Wester SM: Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer 101 (6): 1404-13, 2004.
- Marzano AV, Berti E, Paulli M, et al.: Cytophagic histiocytic panniculitis and subcutaneous panniculitis-like T-cell lymphoma: report of 7 cases. Arch Dermatol 136 (7): 889-96, 2000.
- Hoque SR, Child FJ, Whittaker SJ, et al.: Subcutaneous panniculitis-like T-cell lymphoma: a clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol 148 (3): 516-25, 2003.
- Salhany KE, Macon WR, Choi JK, et al.: Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol 22 (7): 881-93, 1998.
- Massone C, Chott A, Metze D, et al.: Subcutaneous, blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: a morphologic, immunophenotypic, and molecular study of 50 patients. Am J Surg Pathol 28 (6): 719-35, 2004.
- Arnulf B, Copie-Bergman C, Delfau-Larue MH, et al.: Nonhepatosplenic gammadelta T-cell lymphoma: a subset of cytotoxic lymphomas with mucosal or skin localization. Blood 91 (5): 1723-31, 1998.
- Toro JR, Liewehr DJ, Pabby N, et al.: Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood 101 (9): 3407-12, 2003.
- Perry AM, Warnke RA, Hu Q, et al.: Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood 122 (22): 3599-606, 2013.
- Egan LJ, Walsh SV, Stevens FM, et al.: Celiac-associated lymphoma. A single institution experience of 30 cases in the combination chemotherapy era. J Clin Gastroenterol 21 (2): 123-9, 1995.
- Gale J, Simmonds PD, Mead GM, et al.: Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol 18 (4): 795-803, 2000.
- Di Sabatino A, Biagi F, Gobbi PG, et al.: How I treat enteropathy-associated T-cell lymphoma. Blood 119 (11): 2458-68, 2012.
- Daum S, Ullrich R, Heise W, et al.: Intestinal non-Hodgkin's lymphoma: a multicenter prospective clinical study from the German Study Group on Intestinal non-Hodgkin's Lymphoma. J Clin Oncol 21 (14): 2740-6, 2003.
- Sieniawski M, Angamuthu N, Boyd K, et al.: Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood 115 (18): 3664-70, 2010.
- Shimada K, Matsue K, Yamamoto K, et al.: Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol 26 (19): 3189-95, 2008.
- Ponzoni M, Ferreri AJ, Campo E, et al.: Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol 25 (21): 3168-73, 2007.
- Shimada K, Yamaguchi M, Atsuta Y, et al.: Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone combined with high-dose methotrexate plus intrathecal chemotherapy for newly diagnosed intravascular large B-cell lymphoma (PRIMEUR-IVL): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21 (4): 593-602, 2020.
- Blum KA, Lozanski G, Byrd JC: Adult Burkitt leukemia and lymphoma. Blood 104 (10): 3009-20, 2004.
- Onciu M, Schlette E, Zhou Y, et al.: Secondary chromosomal abnormalities predict outcome in pediatric and adult high-stage Burkitt lymphoma. Cancer 107 (5): 1084-92, 2006.
- Macpherson N, Lesack D, Klasa R, et al.: Small noncleaved, non-Burkitt's (Burkit-Like) lymphoma: cytogenetics predict outcome and reflect clinical presentation. J Clin Oncol 17 (5): 1558-67, 1999.
- Dave SS, Fu K, Wright GW, et al.: Molecular diagnosis of Burkitt's lymphoma. N Engl J Med 354 (23): 2431-42, 2006.
- Hummel M, Bentink S, Berger H, et al.: A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med 354 (23): 2419-30, 2006.
- Salaverria I, Siebert R: The gray zone between Burkitt's lymphoma and diffuse large B-cell lymphoma from a genetics perspective. J Clin Oncol 29 (14): 1835-43, 2011.
- Thomas DA, Faderl S, O'Brien S, et al.: Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 106 (7): 1569-80, 2006.
- Dunleavy K, Pittaluga S, Shovlin M, et al.: Low-intensity therapy in adults with Burkitt's lymphoma. N Engl J Med 369 (20): 1915-25, 2013.
- Hoelzer D, Walewski J, Döhner H, et al.: Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood 124 (26): 3870-9, 2014.
- Ribrag V, Koscielny S, Bosq J, et al.: Rituximab and dose-dense chemotherapy for adults with Burkitt's lymphoma: a randomised, controlled, open-label, phase 3 trial. Lancet 387 (10036): 2402-11, 2016.
- Magrath I, Adde M, Shad A, et al.: Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol 14 (3): 925-34, 1996.
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
- Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001.
- Mead GM, Sydes MR, Walewski J, et al.: An international evaluation of CODOX-M and CODOX-M alternating with IVAC in adult Burkitt's lymphoma: results of United Kingdom Lymphoma Group LY06 study. Ann Oncol 13 (8): 1264-74, 2002.
- Rizzieri DA, Johnson JL, Niedzwiecki D, et al.: Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: final results of Cancer and Leukemia Group B Study 9251. Cancer 100 (7): 1438-48, 2004.
- Noy A, Lee JY, Cesarman E, et al.: AMC 048: modified CODOX-M/IVAC-rituximab is safe and effective for HIV-associated Burkitt lymphoma. Blood 126 (2): 160-6, 2015.
- Morel P, Lepage E, Brice P, et al.: Prognosis and treatment of lymphoblastic lymphoma in adults: a report on 80 patients. J Clin Oncol 10 (7): 1078-85, 1992.
- Verdonck LF, Dekker AW, de Gast GC, et al.: Autologous bone marrow transplantation for adult poor-risk lymphoblastic lymphoma in first remission. J Clin Oncol 10 (4): 644-6, 1992.
- Thomas DA, O'Brien S, Cortes J, et al.: Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 104 (6): 1624-30, 2004.
- Sweetenham JW, Santini G, Qian W, et al.: High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 19 (11): 2927-36, 2001.
- Höllsberg P, Hafler DA: Seminars in medicine of the Beth Israel Hospital, Boston. Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med 328 (16): 1173-82, 1993.
- Foss FM, Aquino SL, Ferry JA: Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 10-2003. A 72-year-old man with rapidly progressive leukemia, rash, and multiorgan failure. N Engl J Med 348 (13): 1267-75, 2003.
- Shimoyama M: Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984-87). Br J Haematol 79 (3): 428-37, 1991.
- Takasaki Y, Iwanaga M, Imaizumi Y, et al.: Long-term study of indolent adult T-cell leukemia-lymphoma. Blood 115 (22): 4337-43, 2010.
- Yamada Y, Tomonaga M, Fukuda H, et al.: A new G-CSF-supported combination chemotherapy, LSG15, for adult T-cell leukaemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haematol 113 (2): 375-82, 2001.
- Fukushima T, Miyazaki Y, Honda S, et al.: Allogeneic hematopoietic stem cell transplantation provides sustained long-term survival for patients with adult T-cell leukemia/lymphoma. Leukemia 19 (5): 829-34, 2005.
- Katsuya H, Yamanaka T, Ishitsuka K, et al.: Prognostic index for acute- and lymphoma-type adult T-cell leukemia/lymphoma. J Clin Oncol 30 (14): 1635-40, 2012.
- Itonaga H, Tsushima H, Taguchi J, et al.: Treatment of relapsed adult T-cell leukemia/lymphoma after allogeneic hematopoietic stem cell transplantation: the Nagasaki Transplant Group experience. Blood 121 (1): 219-25, 2013.
- Ishida T, Hishizawa M, Kato K, et al.: Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood 120 (8): 1734-41, 2012.
- Katsuya H, Ishitsuka K, Utsunomiya A, et al.: Treatment and survival among 1594 patients with ATL. Blood 126 (24): 2570-7, 2015.
- Gill PS, Harrington W, Kaplan MH, et al.: Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med 332 (26): 1744-8, 1995.
- Matutes E, Taylor GP, Cavenagh J, et al.: Interferon alpha and zidovudine therapy in adult T-cell leukaemia lymphoma: response and outcome in 15 patients. Br J Haematol 113 (3): 779-84, 2001.
- Hermine O, Allard I, Lévy V, et al.: A prospective phase II clinical trial with the use of zidovudine and interferon-alpha in the acute and lymphoma forms of adult T-cell leukemia/lymphoma. Hematol J 3 (6): 276-82, 2002.
- Bazarbachi A, Plumelle Y, Carlos Ramos J, et al.: Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol 28 (27): 4177-83, 2010.
- Bazarbachi A, Suarez F, Fields P, et al.: How I treat adult T-cell leukemia/lymphoma. Blood 118 (7): 1736-45, 2011.
- Ishida T, Fujiwara H, Nosaka K, et al.: Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATLL-002. J Clin Oncol 34 (34): 4086-4093, 2016.
- Simone CB, Morris JC, Stewart DM, et al.: Radiation therapy for the management of patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Blood 120 (9): 1816-9, 2012.
- Ureshino H, Kamachi K, Kimura S: Mogamulizumab for the Treatment of Adult T-cell Leukemia/Lymphoma. Clin Lymphoma Myeloma Leuk 19 (6): 326-331, 2019.
- Cohen JB, Zain JM, Kahl BS: Current Approaches to Mantle Cell Lymphoma: Diagnosis, Prognosis, and Therapies. Am Soc Clin Oncol Educ Book 37: 512-525, 2017.
- Clot G, Jares P, Giné E, et al.: A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 132 (4): 413-422, 2018.
- Greenwell IB, Staton AD, Lee MJ, et al.: Complex karyotype in patients with mantle cell lymphoma predicts inferior survival and poor response to intensive induction therapy. Cancer 124 (11): 2306-2315, 2018.
- Dreyling M, Klapper W, Rule S: Blastoid and pleomorphic mantle cell lymphoma: still a diagnostic and therapeutic challenge! Blood 132 (26): 2722-2729, 2018.
- Jain P, Dreyling M, Seymour JF, et al.: High-Risk Mantle Cell Lymphoma: Definition, Current Challenges, and Management. J Clin Oncol 38 (36): 4302-4316, 2020.
- Herrmann A, Hoster E, Zwingers T, et al.: Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol 27 (4): 511-8, 2009.
- Majlis A, Pugh WC, Rodriguez MA, et al.: Mantle cell lymphoma: correlation of clinical outcome and biologic features with three histologic variants. J Clin Oncol 15 (4): 1664-71, 1997.
- Tiemann M, Schrader C, Klapper W, et al.: Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 131 (1): 29-38, 2005.
- Campo E, Raffeld M, Jaffe ES: Mantle-cell lymphoma. Semin Hematol 36 (2): 115-27, 1999.
- Martin P, Chadburn A, Christos P, et al.: Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 27 (8): 1209-13, 2009.
- Cohen JB, Han X, Jemal A, et al.: Deferred therapy is associated with improved overall survival in patients with newly diagnosed mantle cell lymphoma. Cancer 122 (15): 2356-63, 2016.
- Gerson JN, Handorf E, Villa D, et al.: Survival Outcomes of Younger Patients With Mantle Cell Lymphoma Treated in the Rituximab Era. J Clin Oncol 37 (6): 471-480, 2019.
- Goy A, Kalayoglu Besisik S, Drach J, et al.: Longer-term follow-up and outcome by tumour cell proliferation rate (Ki-67) in patients with relapsed/refractory mantle cell lymphoma treated with lenalidomide on MCL-001(EMERGE) pivotal trial. Br J Haematol 170 (4): 496-503, 2015.
- Ruan J, Martin P, Shah B, et al.: Lenalidomide plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. N Engl J Med 373 (19): 1835-44, 2015.
- Wang ML, Rule S, Martin P, et al.: Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369 (6): 507-16, 2013.
- Wang ML, Blum KA, Martin P, et al.: Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood 126 (6): 739-45, 2015.
- Ruan J, Martin P, Christos P, et al.: Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood 132 (19): 2016-2025, 2018.
- Jain P, Zhao S, Lee HJ, et al.: Ibrutinib With Rituximab in First-Line Treatment of Older Patients With Mantle Cell Lymphoma. J Clin Oncol 40 (2): 202-212, 2022.
- Wang ML, Jain P, Zhao S, et al.: Ibrutinib-rituximab followed by R-HCVAD as frontline treatment for young patients (≤65 years) with mantle cell lymphoma (WINDOW-1): a single-arm, phase 2 trial. Lancet Oncol 23 (3): 406-415, 2022.
- Giné E, de la Cruz F, Jiménez Ubieto A, et al.: Ibrutinib in Combination With Rituximab for Indolent Clinical Forms of Mantle Cell Lymphoma (IMCL-2015): A Multicenter, Open-Label, Single-Arm, Phase II Trial. J Clin Oncol 40 (11): 1196-1205, 2022.
- Dreyling M, Jurczak W, Jerkeman M, et al.: Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 387 (10020): 770-8, 2016.
- Tam CS, Anderson MA, Pott C, et al.: Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N Engl J Med 378 (13): 1211-1223, 2018.
- Wang ML, Jurczak W, Jerkeman M, et al.: Ibrutinib plus Bendamustine and Rituximab in Untreated Mantle-Cell Lymphoma. N Engl J Med 386 (26): 2482-2494, 2022.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al.: Treatment of Older Patients With Mantle Cell Lymphoma (MCL): Long-Term Follow-Up of the Randomized European MCL Elderly Trial. J Clin Oncol 38 (3): 248-256, 2020.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Robak T, Jin J, Pylypenko H, et al.: Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol 19 (11): 1449-1458, 2018.
- Hermine O, Hoster E, Walewski J, et al.: Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 388 (10044): 565-75, 2016.
- Hermine O, Jiang L, Walewski J, et al.: Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL younger): a long-term follow-up of the randomized, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. [Abstract] Blood 138 (Suppl 1); A-380, 2021.
- Hermine O, Jiang L, Walewski J, et al.: High-Dose Cytarabine and Autologous Stem-Cell Transplantation in Mantle Cell Lymphoma: Long-Term Follow-Up of the Randomized Mantle Cell Lymphoma Younger Trial of the European Mantle Cell Lymphoma Network. J Clin Oncol 41 (3): 479-484, 2023.
- Khouri IF, Lee MS, Saliba RM, et al.: Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 21 (23): 4407-12, 2003.
- Dreyling M, Lenz G, Hoster E, et al.: Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 105 (7): 2677-84, 2005.
- Geisler CH, Kolstad A, Laurell A, et al.: Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 112 (7): 2687-93, 2008.
- Tam CS, Bassett R, Ledesma C, et al.: Mature results of the M. D. Anderson Cancer Center risk-adapted transplantation strategy in mantle cell lymphoma. Blood 113 (18): 4144-52, 2009.
- Damon LE, Johnson JL, Niedzwiecki D, et al.: Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 27 (36): 6101-8, 2009.
- Fenske TS, Zhang MJ, Carreras J, et al.: Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 32 (4): 273-81, 2014.
- Le Gouill S, Thieblemont C, Oberic L, et al.: Rituximab after Autologous Stem-Cell Transplantation in Mantle-Cell Lymphoma. N Engl J Med 377 (13): 1250-1260, 2017.
- Wang M, Fayad L, Wagner-Bartak N, et al.: Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 13 (7): 716-23, 2012.
- Trněný M, Lamy T, Walewski J, et al.: Lenalidomide versus investigator's choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): a phase 2, randomised, multicentre trial. Lancet Oncol 17 (3): 319-31, 2016.
- Wang M, Rule S, Zinzani PL, et al.: Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 391 (10121): 659-667, 2018.
- Song Y, Zhou K, Zou D, et al.: Zanubrutinib in relapsed/refractory mantle cell lymphoma: long-term efficacy and safety results from a phase 2 study. Blood 139 (21): 3148-3158, 2022.
- Wang M, Munoz J, Goy A, et al.: Three-Year Follow-Up of KTE-X19 in Patients With Relapsed/Refractory Mantle Cell Lymphoma, Including High-Risk Subgroups, in the ZUMA-2 Study. J Clin Oncol 41 (3): 555-567, 2023.
- Martin P, Ruan J, Leonard JP: The potential for chemotherapy-free strategies in mantle cell lymphoma. Blood 130 (17): 1881-1888, 2017.
- Morrison VA, Dunn DL, Manivel JC, et al.: Clinical characteristics of post-transplant lymphoproliferative disorders. Am J Med 97 (1): 14-24, 1994.
- Knowles DM, Cesarman E, Chadburn A, et al.: Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 85 (2): 552-65, 1995.
- Leblond V, Dhedin N, Mamzer Bruneel MF, et al.: Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol 19 (3): 772-8, 2001.
- Ghobrial IM, Habermann TM, Maurer MJ, et al.: Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol 23 (30): 7574-82, 2005.
- Evens AM, David KA, Helenowski I, et al.: Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol 28 (6): 1038-46, 2010.
- Dierickx D, Tousseyn T, Gheysens O: How I treat posttransplant lymphoproliferative disorders. Blood 126 (20): 2274-83, 2015.
- Kuehnle I, Huls MH, Liu Z, et al.: CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood 95 (4): 1502-5, 2000.
- Trappe RU, Dierickx D, Zimmermann H, et al.: Response to Rituximab Induction Is a Predictive Marker in B-Cell Post-Transplant Lymphoproliferative Disorder and Allows Successful Stratification Into Rituximab or R-CHOP Consolidation in an International, Prospective, Multicenter Phase II Trial. J Clin Oncol 35 (5): 536-543, 2017.
- Leblond V, Sutton L, Dorent R, et al.: Lymphoproliferative disorders after organ transplantation: a report of 24 cases observed in a single center. J Clin Oncol 13 (4): 961-8, 1995.
- Mamzer-Bruneel MF, Lomé C, Morelon E, et al.: Durable remission after aggressive chemotherapy for very late post-kidney transplant lymphoproliferation: A report of 16 cases observed in a single center. J Clin Oncol 18 (21): 3622-32, 2000.
- Swinnen LJ: Durable remission after aggressive chemotherapy for post-cardiac transplant lymphoproliferation. Leuk Lymphoma 28 (1-2): 89-101, 1997.
- McCarthy M, Ramage J, McNair A, et al.: The clinical diversity and role of chemotherapy in lymphoproliferative disorder in liver transplant recipients. J Hepatol 27 (6): 1015-21, 1997.
- Leblond V, Davi F, Charlotte F, et al.: Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol 16 (6): 2052-9, 1998.
- Senderowicz AM, Vitetta E, Headlee D, et al.: Complete sustained response of a refractory, post-transplantation, large B-cell lymphoma to an anti-CD22 immunotoxin. Ann Intern Med 126 (11): 882-5, 1997.
- Haddad E, Paczesny S, Leblond V, et al.: Treatment of B-lymphoproliferative disorder with a monoclonal anti-interleukin-6 antibody in 12 patients: a multicenter phase 1-2 clinical trial. Blood 97 (6): 1590-7, 2001.
- Soslow RA, Davis RE, Warnke RA, et al.: True histiocytic lymphoma following therapy for lymphoblastic neoplasms. Blood 87 (12): 5207-12, 1996.
- Kamel OW, Gocke CD, Kell DL, et al.: True histiocytic lymphoma: a study of 12 cases based on current definition. Leuk Lymphoma 18 (1-2): 81-6, 1995.
- Nador RG, Cesarman E, Chadburn A, et al.: Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88 (2): 645-56, 1996.
- Shimada K, Hayakawa F, Kiyoi H: Biology and management of primary effusion lymphoma. Blood 132 (18): 1879-1888, 2018.
- Castillo JJ, Bibas M, Miranda RN: The biology and treatment of plasmablastic lymphoma. Blood 125 (15): 2323-30, 2015.
- Al-Malki MM, Castillo JJ, Sloan JM, et al.: Hematopoietic cell transplantation for plasmablastic lymphoma: a review. Biol Blood Marrow Transplant 20 (12): 1877-84, 2014.
- Cattaneo C, Re A, Ungari M, et al.: Plasmablastic lymphoma among human immunodeficiency virus-positive patients: results of a single center's experience. Leuk Lymphoma 56 (1): 267-9, 2015.
Stage Information for NHL
Stage is important in selecting a treatment for patients with non-Hodgkin lymphoma (NHL). Chest and abdominal computed tomography (CT) scans are usually part of the staging evaluation for all lymphoma patients. The staging system is similar to the staging system used for Hodgkin lymphoma (HL).
Common among patients with NHL is involvement of the following:
- Noncontiguous lymph nodes.
- Waldeyer ring.
- Epitrochlear nodes.
- Gastrointestinal tract.
- Extranodal presentations. (A single extranodal site is occasionally the only site of involvement in patients with diffuse lymphoma.)
- Bone marrow.
- Liver (especially common in patients with low-grade lymphomas).
Cytological examination of cerebrospinal fluid may be positive in patients with aggressive NHL. Involvement of hilar and mediastinal lymph nodes is less common than in HL. Mediastinal adenopathy, however, is a prominent feature of lymphoblastic lymphoma and primary mediastinal B-cell lymphoma, entities primarily found in young adults.
Most patients with NHL present with advanced (stage III or stage IV) disease that can often be identified with limited staging procedures such as CT scanning and biopsies of the bone marrow and other accessible sites of involvement. In a retrospective review of over 32,000 cases of lymphoma in France, up to 40% of diagnoses were made by core needle biopsy, and 60% were made by excisional biopsy.[1] After expert review, core needle biopsy provided a definite diagnosis in 92.3% of cases, but it was less conclusive than excisional biopsy, which provided a definite diagnosis in 98.1% of cases (P < .0001). Laparoscopic biopsy or laparotomy is not required for staging but rarely may be necessary to establish a diagnosis or histological type.[2] Positron emission tomography (PET) with fluorine F 18-fludeoxyglucose can be used for initial staging and for follow-up after therapy as a supplement to CT scanning.[3] Interim PET scans after two to four cycles of therapy did not provide reliable prognostic information because of problems of interobserver reproducibility in a large cooperative group trial (ECOG-E344 [NCT00274924]) and lack of difference in outcome between PET-negative and PET-positive/biopsy-negative patients in two prospective trials [4,5,6] and in a meta-analysis.[7] For patients with follicular lymphoma, a positive PET result after therapy has a worse prognosis; however, it is unclear whether a positive PET result is predictive when further or different therapy is implemented.[8]
In a retrospective study of 130 patients with diffuse large B-cell lymphoma, PET scanning identified all clinically important marrow involvement from lymphoma, and bone marrow biopsy did not upstage any patient's lymphoma.[9] A retrospective study of 580 patients with follicular lymphoma from seven National Cancer Institute-sponsored trials showed no improvement in assessing response to therapy when bone marrow biopsy was added to radiological imaging.[10] The workup of NHL should include bone marrow biopsy when management would change (e.g., determining limited stage vs. advanced stage) or when evaluating cytopenias.
Staging Subclassification System
Lugano Classification
The American Joint Committee on Cancer (AJCC) has adopted the Lugano classification to evaluate and stage lymphoma.[11] The Lugano classification system replaces the Ann Arbor classification system, which was adopted in 1971 at the Ann Arbor Conference,[12] with some modifications 18 years later from the Cotswolds meeting.[13,14]
Table 2. Lugano Classification for Hodgkin and Non-Hodgkin Lymphomaa| Stage | Stage Description | Illustration |
|---|
| CSF = cerebrospinal fluid; CT = computed tomography; DLBCL = diffuse large B-cell lymphoma; NHL = non-Hodgkin lymphoma. |
| a Hodgkin and Non-Hodgkin Lymphomas. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp. 937–58. |
| b Stage II bulky may be considered either early or advanced stage based on lymphoma histology and prognostic factors. |
| c The definition of disease bulk varies according to lymphoma histology. In the Lugano classification, bulk ln Hodgkin lymphoma is defined as a mass greater than one-third of the thoracic diameter on CT of the chest or a mass >10 cm. For NHL, the recommended definitions of bulk vary by lymphoma histology. In follicular lymphoma, 6 cm has been suggested based on the Follicular Lymphoma International Prognostic Index-2 and its validation. In DLBCL, cutoffs ranging from 5 cm to 10 cm have been used, although 10 cm is recommended. |
| Limited stage |
| I | Involvement of a single lymphatic site (i.e., nodal region, Waldeyer's ring, thymus, or spleen). |
|
| IE | Single extralymphatic site in the absence of nodal involvement (rare in Hodgkin lymphoma). | |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm. |
|
| IIE | Contiguous extralymphatic extension from a nodal site with or without involvement of other lymph node regions on the same side of the diaphragm. |
|
| II bulkyb | Stage II with disease bulk.c | |
| Advanced stage |
| III | Involvement of lymph node regions on both sides of the diaphragm; nodes above the diaphragm with spleen involvement. |
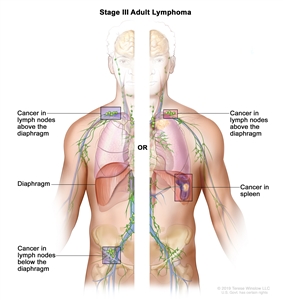
|
| IV | Diffuse or disseminated involvement of one or more extralymphatic organs, with or without associated lymph node involvement; or noncontiguous extralymphatic organ involvement in conjunction with nodal stage II disease; or any extralymphatic organ involvement in nodal stage III disease. Stage IV includes any involvement of the CSF, bone marrow, liver, or multiple lung lesions (other than by direct extension in stage IIE disease). |
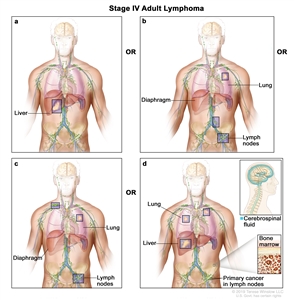
|
| Note: Hodgkin lymphoma uses A or B designation with stage group. A/B is no longer used in NHL. |
Occasionally, specialized staging systems are used. The physician should be aware of the system used in a specific report.
The E designation is used when extranodal lymphoid malignancies arise in tissues separate from, but near, the major lymphatic aggregates. Stage IV refers to disease that is diffusely spread throughout an extranodal site, such as the liver. If pathological proof of involvement of one or more extralymphatic sites has been documented, the symbol for the site of involvement, followed by a plus sign (+), is listed.
Table 3. Notation to Identify Specific Sites| N = nodes | H = liver | L = lung | M = bone marrow |
| S = spleen | P = pleura | O = bone | D = skin |
Current practice assigns a clinical stage based on the findings of the clinical evaluation and a pathological stage based on the findings made as a result of invasive procedures beyond the initial biopsy.
For example, on percutaneous biopsy, a patient with inguinal adenopathy and a positive lymphangiogram without systemic symptoms might have involvement of the liver and bone marrow. The precise stage of such a patient would be clinical stage IIA, pathological stage IVA(H+)(M+).
Several other factors that are not included in the above staging system are important for the staging and prognosis of patients with NHL. These factors include the following:
- Age.
- Performance status (PS).
- Tumor size.
- Lactate dehydrogenase (LDH) values.
- The number of extranodal sites.
The National Comprehensive Cancer Network International Prognostic Index (IPI) for aggressive NHL (diffuse large cell lymphoma) identifies the following five significant risk factors prognostic of overall survival (OS) and their associated risk scores:[15]
- Age.
- <40 years: 0.
- 41–60 years: 1.
- 61–75 years: 2.
- >75 years: 3.
- Stage III/IV: 1.
- PS 2/3/4: 1.
- Serum LDH.
- Normalized: 0.
- >1x–3x: 1.
- >3x: 2.
- Number of extranodal sites ≥2: 1.
Risk scores:
- Low (0 or 1): 5-year OS rate, 96%; progression-free survival (PFS) rate, 91%.
- Low intermediate (2 or 3): 5-year OS rate, 82%; PFS rate, 74%.
- High intermediate (4 or 5): 5-year OS rate, 64%; PFS rate, 51%.
- High (>6): 5-year OS rate, 33%; PFS rate, 30%.
Age-adjusted and stage-adjusted modifications of this IPI are used for younger patients with localized disease.[16] Shorter intervals of time between diagnosis and treatment appear to be a surrogate for poor prognostic biological factors.[17]
The BCL2 gene and rearrangement of the MYC gene or dual overexpression of the MYC gene, or both, confer a particularly poor prognosis.[18,19] Patients at high risk of relapse may benefit from consolidation therapy or other approaches under clinical evaluation.[20] Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.[21]
References:
- Syrykh C, Chaouat C, Poullot E, et al.: Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 140 (24): 2573-2583, 2022.
- Mann GB, Conlon KC, LaQuaglia M, et al.: Emerging role of laparoscopy in the diagnosis of lymphoma. J Clin Oncol 16 (5): 1909-15, 1998.
- Barrington SF, Mikhaeel NG, Kostakoglu L, et al.: Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 32 (27): 3048-58, 2014.
- Horning SJ, Juweid ME, Schöder H, et al.: Interim positron emission tomography scans in diffuse large B-cell lymphoma: an independent expert nuclear medicine evaluation of the Eastern Cooperative Oncology Group E3404 study. Blood 115 (4): 775-7; quiz 918, 2010.
- Moskowitz CH, Schöder H, Teruya-Feldstein J, et al.: Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 28 (11): 1896-903, 2010.
- Pregno P, Chiappella A, Bellò M, et al.: Interim 18-FDG-PET/CT failed to predict the outcome in diffuse large B-cell lymphoma patients treated at the diagnosis with rituximab-CHOP. Blood 119 (9): 2066-73, 2012.
- Sun N, Zhao J, Qiao W, et al.: Predictive value of interim PET/CT in DLBCL treated with R-CHOP: meta-analysis. Biomed Res Int 2015: 648572, 2015.
- Pyo J, Won Kim K, Jacene HA, et al.: End-therapy positron emission tomography for treatment response assessment in follicular lymphoma: a systematic review and meta-analysis. Clin Cancer Res 19 (23): 6566-77, 2013.
- Khan AB, Barrington SF, Mikhaeel NG, et al.: PET-CT staging of DLBCL accurately identifies and provides new insight into the clinical significance of bone marrow involvement. Blood 122 (1): 61-7, 2013.
- Rutherford SC, Yin J, Pederson L, et al.: Relevance of Bone Marrow Biopsies for Response Assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol 41 (2): 336-342, 2023.
- Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
- Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin's Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971.
- Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989.
- National Cancer Institute sponsored study of classifications of non-Hodgkin's lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin's Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Scott DW, King RL, Staiger AM, et al.: High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131 (18): 2060-2064, 2018.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- A predictive model for aggressive non-Hodgkin's lymphoma. The International Non-Hodgkin's Lymphoma Prognostic Factors Project. N Engl J Med 329 (14): 987-94, 1993.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
Treatment Option Overview for NHL
Treatment of non-Hodgkin lymphoma (NHL) depends on the histological type and stage. Many of the improvements in survival have been made using clinical trials (experimental therapy) that have attempted to improve on the best available accepted therapy (conventional or standard therapy).
In asymptomatic patients with indolent forms of advanced NHL, treatment may be deferred until the patient becomes symptomatic as the disease progresses. When treatment is deferred, the clinical course of patients with indolent NHL varies; frequent and careful observation is required so that effective treatment can be initiated when the clinical course of the disease accelerates. Some patients have a prolonged indolent course, but others have disease that rapidly evolves into more aggressive types of NHL that require immediate treatment.
Radiation techniques differ somewhat from those used in the treatment of Hodgkin lymphoma. The dose of radiation therapy usually varies from 25 Gy to 50 Gy and is dependent on factors that include the histological type of lymphoma, the patient's stage and overall condition, the goal of treatment (curative or palliative), the proximity of sensitive surrounding organs, and whether the patient is being treated with radiation therapy alone or in combination with chemotherapy. Given the patterns of disease presentations and relapse, treatment may need to include unusual sites such as Waldeyer ring, epitrochlear nodes, or mesenteric nodes. The associated morbidity of the treatment must be considered carefully. Most patients who receive radiation are treated on only one side of the diaphragm. Localized presentations of extranodal NHL may be treated with involved-field techniques with significant (>50%) success.
Table 4. Treatment Options for Non-Hodgkin Lymphoma (NHL)| Stage | Treatment Options |
|---|
| BMT = bone marrow transplantation; CAR = chimeric antigen receptor; CNS = central nervous system; IF-XRT = involved-field radiation therapy; PI3K = phosphatidylinositol 3-kinase; R-ACVBP = rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; SCT = stem cell transplantation. |
| Indolent Stage I and Indolent, Contiguous Stage II NHL | Radiation therapy |
| Rituximab with or without chemotherapy |
| Watchful waiting |
| Other therapies as designated for patients with advanced-stage disease |
| Indolent, Noncontiguous Stage II/III/IV NHL | Watchful waiting for asymptomatic patients |
| Rituximab alone or in combination with cytotoxic agents used in front-line therapy |
| Lenalidomide and rituximab |
| Maintenance rituximab |
| Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy |
| PI3K inhibitor |
| EZH2 inhibitor |
| Radiolabeled anti-CD20 monoclonal antibodies |
| Intensive therapy with chemotherapy with or without total-body irradiation or high-dose radioimmunotherapy followed by autologous or allogeneic BMT or peripheral SCT (under clinical evaluation) |
| Phase III clinical trials comparing chemotherapy alone versus chemotherapy followed by anti-idiotype vaccine |
| Extended-field radiation therapy (stage III patients only) (under clinical evaluation) |
| Ofatumumab (under clinical evaluation) |
| Short-course low-dose, palliative radiation therapy (2 × 2 Gy) (under clinical evaluation) |
| Indolent, Recurrent NHL | Rituximab alone or in combination with cytotoxic agents used in front-line therapy |
| Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy |
| Lenalidomide and rituximab |
| PI3K inhibitor |
| EZH2 inhibitor |
| Palliative radiation therapy |
| Chemotherapy (single agent or combination) |
| Radiolabeled anti-CD20 monoclonal antibodies |
| CAR T-cell therapy |
| Bispecific T-cell engagers |
| SCT |
| Aggressive Stage I and Aggressive, Contiguous Stage II NHL | R-CHOP with or without IF-XRT |
| R-ACVBP (under clinical evaluation) |
| Aggressive, Noncontiguous Stage II/III/IV NHL | R-CHOP |
| Other combination chemotherapy |
| BMT or SCT(under clinical evaluation) |
| Radiation therapy consolidation to sites of bulky disease(under clinical evaluation) |
| Lymphoblastic Lymphoma/Acute Lymphocytic Leukemia | Intensive therapy |
| Radiation therapy |
| Diffuse, Small, Noncleaved-Cell/Burkitt Lymphoma | Aggressive multidrug regimens |
| CNS prophylaxis |
| Aggressive, Recurrent NHL | CAR T-cell therapy for primary refractory disease or relapse within one year |
| BMT or SCT consolidation |
| CAR T-cell therapy for relapse after autologous SCT |
| Tafasitamab plus lenalidomide |
| Rituximab plus lenalidomide |
| Polatuzumab vedotin plus rituximab and bendamustine |
| Loncastuximab tesirine |
| Bispecific T-cell engagers |
| Palliative radiation therapy |
Even though existing treatments cure a significant fraction of patients with lymphoma, numerous clinical trials that explore treatment improvements are in progress. If possible, patients can be included in these studies. Standardized guidelines for response assessment have been suggested for use in clinical trials.[1]
Several retrospective reviews suggest that routine surveillance scans offer little to no value in patients with diffuse-large B-cell lymphoma (DLBCL) who have attained a clinical complete remission after induction therapy. Prognostic value is also difficult to identify for an interim positron emission tomography-computed tomography scan during induction therapy for DLBCL.[2,3,4,5]
Aggressive lymphomas are increasingly seen in HIV-positive patients. Treatment of these patients requires special consideration. For more information, see AIDS-Related Lymphoma Treatment.
In addition to screening for HIV among patients with aggressive lymphomas, active hepatitis B or hepatitis C can be assessed before treatment with rituximab and/or chemotherapy.[6,7] Patients with detectable hepatitis B viral loads benefit from prophylaxis with entecavir in the context of rituximab therapy.[8,9] Patients with a resolved hepatitis B virus (HBV) infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. Prophylactic nucleoside therapy lowered HBV reactivation from 10.8% to 2.1% in a retrospective study of 326 patients.[10] Similarly, prophylaxis for herpes zoster with acyclovir or valacyclovir and prophylaxis for pneumocystis with trimethoprim/sulfamethoxazole or dapsone are usually applied with rituximab with or without combination chemotherapy. Long-term impaired immune health was evaluated in a retrospective cohort study of 21,690 survivors of DLBCL from the California Cancer Registry. Elevated incidence rate ratios were found up to 10 years later for pneumonia (10.8-fold), meningitis (5.3-fold), immunoglobulin deficiency (17.6-fold), and autoimmune cytopenias (12-fold).[11]
Among 2,508 patients in a Danish registry, the incidence of doxorubicin-induced congestive heart failure increased for 115 NHL survivors with a history of cardiac disease (hazard ratio [HR], 2.71; 95% confidence interval [CI], 1.15−6.36) and/or multiple cardiovascular risk factors (HR, 2.86; 95% CI, 1.56−5.23).[12]
Several unusual presentations of lymphoma occur that often require somewhat modified approaches to staging and therapy. The reader is referred to reviews for a more detailed description of extranodal presentations in the gastrointestinal system,[13,14,15,16,17,18,19,20,21] thyroid,[22,23] spleen,[24] testis,[25,26,27] paranasal sinuses,[28,29,30,31] bone,[32,33] orbit,[34,35,36,37,38] and skin.[39,40,41,42,43,44,45,46,47,48]
For more information, see Primary CNS Lymphoma Treatment.
Castleman Disease
A biopsy of localized or multifocal collections of lymph nodes may lead to a diagnosis of Castleman disease (CD), although it is an uncommon diagnosis. Strictly speaking, this is not a lymphoma, nor is it even a malignancy. Yet, many patients with CD may be seen and treated by hematologists or oncologists.
Localized or unicentric CD is usually asymptomatic and occurs in the mediastinum, which is the most common presentation for CD.[49] Watchful waiting, surgery, or radiation therapy can be used for this indolent form. Multicentric CD (30% of CD patients) presents with lymphadenopathy in multiple sites; symptoms such as fever, night sweats, weight loss, and fatigue; and laboratory abnormalities such as anemia, low albumin, elevated C-reactive protein, and high fibrinogen.[49] Multicentric CD (MCD) is subdivided into human herpes virus-8–associated MCD (usually with HIV or with severe immunocompromise) or idiopathic MCD. Cytopenias and cytokine storm are attributed to interleukin-6 (IL-6) overproduction. MCD is a feature seen in POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin abnormalities) syndrome [50] and TAFRO (thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly) syndrome.[51,52] Therapy with siltuximab (an anti–IL-6 monoclonal antibody), rituximab (an anti-CD20 monoclonal antibody), or chemotherapeutic agents has been presented in anecdotal nonrandomized series.[53,54,55,56]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Cheson BD, Horning SJ, Coiffier B, et al.: Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol 17 (4): 1244, 1999.
- Mamot C, Klingbiel D, Hitz F, et al.: Final Results of a Prospective Evaluation of the Predictive Value of Interim Positron Emission Tomography in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP-14 (SAKK 38/07). J Clin Oncol 33 (23): 2523-9, 2015.
- Thompson CA, Ghesquieres H, Maurer MJ, et al.: Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol 32 (31): 3506-12, 2014.
- El-Galaly TC, Jakobsen LH, Hutchings M, et al.: Routine Imaging for Diffuse Large B-Cell Lymphoma in First Complete Remission Does Not Improve Post-Treatment Survival: A Danish-Swedish Population-Based Study. J Clin Oncol 33 (34): 3993-8, 2015.
- Huntington SF, Svoboda J, Doshi JA: Cost-effectiveness analysis of routine surveillance imaging of patients with diffuse large B-cell lymphoma in first remission. J Clin Oncol 33 (13): 1467-74, 2015.
- Niitsu N, Hagiwara Y, Tanae K, et al.: Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J Clin Oncol 28 (34): 5097-100, 2010.
- Dong HJ, Ni LN, Sheng GF, et al.: Risk of hepatitis B virus (HBV) reactivation in non-Hodgkin lymphoma patients receiving rituximab-chemotherapy: a meta-analysis. J Clin Virol 57 (3): 209-14, 2013.
- Huang YH, Hsiao LT, Hong YC, et al.: Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 31 (22): 2765-72, 2013.
- Li H, Zhang HM, Chen LF, et al.: Prophylactic lamivudine to improve the outcome of HBsAg-positive lymphoma patients during chemotherapy: a systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 39 (1): 80-92, 2015.
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- Shree T, Li Q, Glaser SL, et al.: Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (15): 1664-1675, 2020.
- Salz T, Zabor EC, de Nully Brown P, et al.: Preexisting Cardiovascular Risk and Subsequent Heart Failure Among Non-Hodgkin Lymphoma Survivors. J Clin Oncol 35 (34): 3837-3843, 2017.
- Maor MH, Velasquez WS, Fuller LM, et al.: Stomach conservation in stages IE and IIE gastric non-Hodgkin's lymphoma. J Clin Oncol 8 (2): 266-71, 1990.
- Salles G, Herbrecht R, Tilly H, et al.: Aggressive primary gastrointestinal lymphomas: review of 91 patients treated with the LNH-84 regimen. A study of the Groupe d'Etude des Lymphomes Agressifs. Am J Med 90 (1): 77-84, 1991.
- Taal BG, Burgers JM, van Heerde P, et al.: The clinical spectrum and treatment of primary non-Hodgkin's lymphoma of the stomach. Ann Oncol 4 (10): 839-46, 1993.
- Tondini C, Giardini R, Bozzetti F, et al.: Combined modality treatment for primary gastrointestinal non-Hodgkin's lymphoma: the Milan Cancer Institute experience. Ann Oncol 4 (10): 831-7, 1993.
- d'Amore F, Brincker H, Grønbaek K, et al.: Non-Hodgkin's lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 12 (8): 1673-84, 1994.
- Haim N, Leviov M, Ben-Arieh Y, et al.: Intermediate and high-grade gastric non-Hodgkin's lymphoma: a prospective study of non-surgical treatment with primary chemotherapy, with or without radiotherapy. Leuk Lymphoma 17 (3-4): 321-6, 1995.
- Koch P, del Valle F, Berdel WE, et al.: Primary gastrointestinal non-Hodgkin's lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 19 (18): 3861-73, 2001.
- Koch P, del Valle F, Berdel WE, et al.: Primary gastrointestinal non-Hodgkin's lymphoma: II. Combined surgical and conservative or conservative management only in localized gastric lymphoma--results of the prospective German Multicenter Study GIT NHL 01/92. J Clin Oncol 19 (18): 3874-83, 2001.
- Koch P, Probst A, Berdel WE, et al.: Treatment results in localized primary gastric lymphoma: data of patients registered within the German multicenter study (GIT NHL 02/96). J Clin Oncol 23 (28): 7050-9, 2005.
- Blair TJ, Evans RG, Buskirk SJ, et al.: Radiotherapeutic management of primary thyroid lymphoma. Int J Radiat Oncol Biol Phys 11 (2): 365-70, 1985.
- Junor EJ, Paul J, Reed NS: Primary non-Hodgkin's lymphoma of the thyroid. Eur J Surg Oncol 18 (4): 313-21, 1992.
- Morel P, Dupriez B, Gosselin B, et al.: Role of early splenectomy in malignant lymphomas with prominent splenic involvement (primary lymphomas of the spleen). A study of 59 cases. Cancer 71 (1): 207-15, 1993.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Liang R, Todd D, Chan TK, et al.: Treatment outcome and prognostic factors for primary nasal lymphoma. J Clin Oncol 13 (3): 666-70, 1995.
- Cheung MM, Chan JK, Lau WH, et al.: Primary non-Hodgkin's lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16 (1): 70-7, 1998.
- Hausdorff J, Davis E, Long G, et al.: Non-Hodgkin's lymphoma of the paranasal sinuses: clinical and pathological features, and response to combined-modality therapy. Cancer J Sci Am 3 (5): 303-11, 1997 Sep-Oct.
- Sasai K, Yamabe H, Kokubo M, et al.: Head-and-neck stages I and II extranodal non-Hodgkin's lymphomas: real classification and selection for treatment modality. Int J Radiat Oncol Biol Phys 48 (1): 153-60, 2000.
- Ferreri AJ, Reni M, Ceresoli GL, et al.: Therapeutic management with adriamycin-containing chemotherapy and radiotherapy of monostotic and polyostotic primary non-Hodgkin's lymphoma of bone in adults. Cancer Invest 16 (8): 554-61, 1998.
- Dubey P, Ha CS, Besa PC, et al.: Localized primary malignant lymphoma of bone. Int J Radiat Oncol Biol Phys 37 (5): 1087-93, 1997.
- Martinet S, Ozsahin M, Belkacémi Y, et al.: Outcome and prognostic factors in orbital lymphoma: a Rare Cancer Network study on 90 consecutive patients treated with radiotherapy. Int J Radiat Oncol Biol Phys 55 (4): 892-8, 2003.
- Uno T, Isobe K, Shikama N, et al.: Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: a multiinstitutional, retrospective review of 50 patients. Cancer 98 (4): 865-71, 2003.
- Sjö LD, Ralfkiaer E, Juhl BR, et al.: Primary lymphoma of the lacrimal sac: an EORTC ophthalmic oncology task force study. Br J Ophthalmol 90 (8): 1004-9, 2006.
- Stefanovic A, Lossos IS: Extranodal marginal zone lymphoma of the ocular adnexa. Blood 114 (3): 501-10, 2009.
- Sjö LD: Ophthalmic lymphoma: epidemiology and pathogenesis. Acta Ophthalmol 87 Thesis 1: 1-20, 2009.
- Geelen FA, Vermeer MH, Meijer CJ, et al.: bcl-2 protein expression in primary cutaneous large B-cell lymphoma is site-related. J Clin Oncol 16 (6): 2080-5, 1998.
- Pandolfino TL, Siegel RS, Kuzel TM, et al.: Primary cutaneous B-cell lymphoma: review and current concepts. J Clin Oncol 18 (10): 2152-68, 2000.
- Sarris AH, Braunschweig I, Medeiros LJ, et al.: Primary cutaneous non-Hodgkin's lymphoma of Ann Arbor stage I: preferential cutaneous relapses but high cure rate with doxorubicin-based therapy. J Clin Oncol 19 (2): 398-405, 2001.
- Grange F, Bekkenk MW, Wechsler J, et al.: Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol 19 (16): 3602-10, 2001.
- Mirza I, Macpherson N, Paproski S, et al.: Primary cutaneous follicular lymphoma: an assessment of clinical, histopathologic, immunophenotypic, and molecular features. J Clin Oncol 20 (3): 647-55, 2002.
- Smith BD, Glusac EJ, McNiff JM, et al.: Primary cutaneous B-cell lymphoma treated with radiotherapy: a comparison of the European Organization for Research and Treatment of Cancer and the WHO classification systems. J Clin Oncol 22 (4): 634-9, 2004.
- Willemze R, Jaffe ES, Burg G, et al.: WHO-EORTC classification for cutaneous lymphomas. Blood 105 (10): 3768-85, 2005.
- El-Helw L, Goodwin S, Slater D, et al.: Primary B-cell lymphoma of the skin: the Sheffield Lymphoma Group Experience (1984-2003). Int J Oncol 25 (5): 1453-8, 2004.
- Zinzani PL, Quaglino P, Pimpinelli N, et al.: Prognostic factors in primary cutaneous B-cell lymphoma: the Italian Study Group for Cutaneous Lymphomas. J Clin Oncol 24 (9): 1376-82, 2006.
- Senff NJ, Noordijk EM, Kim YH, et al.: European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 112 (5): 1600-9, 2008.
- van Rhee F, Voorhees P, Dispenzieri A, et al.: International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 132 (20): 2115-2124, 2018.
- Dispenzieri A: POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am J Hematol 94 (7): 812-827, 2019.
- Zhang Y, Suo SS, Yang HJ, et al.: Clinical features and treatment of 7 Chinese TAFRO syndromes from 96 de novo Castleman diseases: a 10-year retrospective study. J Cancer Res Clin Oncol 146 (2): 357-365, 2020.
- Fujimoto S, Sakai T, Kawabata H, et al.: Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol 94 (9): 975-983, 2019.
- Tonialini L, Bonfichi M, Ferrero S, et al.: Siltuximab in relapsed/refractory multicentric Castleman disease: Experience of the Italian NPP program. Hematol Oncol 36 (4): 689-692, 2018.
- Dong Y, Zhang L, Nong L, et al.: Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 97 (9): 1641-1647, 2018.
- Zhang L, Zhao AL, Duan MH, et al.: Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood 133 (16): 1720-1728, 2019.
- van Rhee F, Wong RS, Munshi N, et al.: Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15 (9): 966-74, 2014.
Treatment of Indolent Stage I and Indolent, Contiguous Stage II NHL
Although localized presentations are uncommon in non-Hodgkin lymphoma (NHL), the goal of treatment is to cure the disease in patients who are shown to have truly localized occurrence after undergoing appropriate staging.
Treatment Options for Indolent Stage I and Indolent, Contiguous Stage II NHL
Treatment options for indolent stage I and indolent, contiguous stage II NHL include the following:
- Radiation therapy.
- Rituximab with or without chemotherapy.
- Watchful waiting.
- Other therapies as designated for patients with advanced-stage disease.
In a prospective randomized trial, 150 patients with stage I or stage II follicular lymphoma were randomly assigned to 30 Gy of involved-field radiation therapy alone or radiation therapy plus six cycles of R-CVP (rituximab, cyclophosphamide, vincristine, prednisolone). With a median follow-up of 9.6 years, the 10-year progression-free survival (PFS) rate favored combined-modality therapy, at 59% (95% confidence interval [CI], 46%–74%) versus 41% for radiation therapy alone (95% CI, 30%–57%) (P = .033). There was no difference in overall survival (OS) (87% and 95%, P = .40).[1][Level of evidence B1]
The National Lymphocare Study identified 471 patients with stage I follicular lymphoma. Of those patients, 206 were rigorously staged with a bone marrow aspirate and biopsy, and computed tomography (CT) scans or positron emission tomography (PET)-CT scans.[2] Nonrandomized treatments included radiation therapy (27%), rituximab-chemotherapy (R-chemotherapy) (28%), watchful waiting (17%), R-chemotherapy plus radiation therapy (13%), and rituximab alone (12%), although more than one-third of the patients started with expectant therapy. With a median follow-up of 57 months, PFS favored R-chemotherapy or R-chemotherapy plus radiation therapy, but OS was nearly identical, all over 90%.[2][Level of evidence C2] Clinical trials are required to answer questions such as the following:[3]
- If the PET-CT scan is clear after excisional biopsy, is watchful waiting or radiation therapy preferred?
- Should rituximab be added to radiation therapy for stage I follicular lymphoma?
- Is there any role for R-chemotherapy plus radiation therapy?
Radiation therapy
Long-term disease control within radiation fields can be achieved in a significant number of patients with indolent stage I or stage II NHL by using dosages of radiation that usually range from 25 Gy to 40 Gy to involved sites or to extended fields that cover adjacent nodal sites.[1,4,5,6] Almost half of all patients treated with radiation therapy alone will relapse out-of-field within 10 years.[1,6,7]
A retrospective review of 512 patients from an international consortium evaluated patients with early-stage follicular lymphoma who received at least 24 Gy of localized radiation therapy at initial presentation. With a median follow-up of 52 months, 29.1% of patients developed recurrent lymphoma at a median of 23 months (range, 1−143 months).[8] With a median follow-up of 33 months after relapse, the 3-year OS rate was 91.4% after patients received subsequent systemic chemotherapy that involved rituximab, usually with chemotherapy.[8]
Very low-dose radiation therapy with 4 Gy (2 Gy × 2 fractions) can result in 50% remission rates for patients who cannot tolerate higher doses.[9] In a multicenter, randomized, prospective trial, 548 patients with follicular or marginal zone lymphoma received radiation therapy, either 4 Gy in 2 fractions or 24 Gy in 12 fractions.[10]
- At a median follow-up of 73.8 months, the 5-year local complete response rate was 89.9% (85.5%–93.1%) after 24 Gy and 70.4% (64.7%–75.4%) after 4 Gy (HR, 3.46; 95% CI, 2.25–5.33, P < .0001).[10]
- Although durable local control was superior for patients who received 24 Gy in 12 fractions, the 4 Gy (2 Gy x 2 fractions) regimen was nearly comparable with less radiation exposure, less time undergoing therapy, and less cost.
In situations in which mediastinal radiation would encompass the left side of the heart or would increase breast cancer risk in young female patients, proton therapy may be considered to reduce the radiation dose to organs at risk.[11]
Rituximab with or without chemotherapy
For symptomatic patients who require therapy, when radiation therapy is contraindicated, or when an alternative treatment is preferred, rituximab with or without chemotherapy can be used (as outlined below for more advanced-stage patients). The value of adjuvant treatment with radiation to decrease relapse, plus rituximab (an anti–CD20 monoclonal antibody) either alone or in combination with chemotherapy, has been extrapolated from trials of patients with advanced-stage disease and has not been confirmed.[12,13]
Watchful waiting
Watchful waiting can be considered for asymptomatic patients.[14] Watchful waiting has never been compared with up-front radiation therapy in a prospective randomized trial; a retrospective analysis of the Surveillance, Epidemiology and End Results (SEER) Program database over 30 years showed improved outcomes for up-front radiation therapy.[15]
Other therapies as designated for patients with advanced-stage disease
Patients with involvement that is not able to be encompassed by radiation therapy are treated as outlined for patients with stage III or stage IV low-grade lymphoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- MacManus M, Fisher R, Roos D, et al.: Randomized Trial of Systemic Therapy After Involved-Field Radiotherapy in Patients With Early-Stage Follicular Lymphoma: TROG 99.03. J Clin Oncol 36 (29): 2918-2925, 2018.
- Friedberg JW, Byrtek M, Link BK, et al.: Effectiveness of first-line management strategies for stage I follicular lymphoma: analysis of the National LymphoCare Study. J Clin Oncol 30 (27): 3368-75, 2012.
- Montoto S: Management of localized-stage follicular lymphoma: changing the paradigm? J Clin Oncol 30 (27): 3328-9, 2012.
- Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003.
- Guckenberger M, Alexandrow N, Flentje M: Radiotherapy alone for stage I-III low grade follicular lymphoma: long-term outcome and comparison of extended field and total nodal irradiation. Radiat Oncol 7: 103, 2012.
- Brady JL, Binkley MS, Hajj C, et al.: Definitive radiotherapy for localized follicular lymphoma staged by 18F-FDG PET-CT: a collaborative study by ILROG. Blood 133 (3): 237-245, 2019.
- Guadagnolo BA, Li S, Neuberg D, et al.: Long-term outcome and mortality trends in early-stage, Grade 1-2 follicular lymphoma treated with radiation therapy. Int J Radiat Oncol Biol Phys 64 (3): 928-34, 2006.
- Binkley MS, Brady JL, Hajj C, et al.: Salvage Treatment and Survival for Relapsed Follicular Lymphoma Following Primary Radiation Therapy: A Collaborative Study on Behalf of ILROG. Int J Radiat Oncol Biol Phys 104 (3): 522-529, 2019.
- Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014.
- Hoskin P, Popova B, Schofield O, et al.: 4 Gy versus 24 Gy radiotherapy for follicular and marginal zone lymphoma (FoRT): long-term follow-up of a multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol 22 (3): 332-340, 2021.
- Dabaja BS, Hoppe BS, Plastaras JP, et al.: Proton therapy for adults with mediastinal lymphomas: the International Lymphoma Radiation Oncology Group guidelines. Blood 132 (16): 1635-1646, 2018.
- Kelsey SM, Newland AC, Hudson GV, et al.: A British National Lymphoma Investigation randomised trial of single agent chlorambucil plus radiotherapy versus radiotherapy alone in low grade, localised non-Hodgkins lymphoma. Med Oncol 11 (1): 19-25, 1994.
- Seymour JF, Pro B, Fuller LM, et al.: Long-term follow-up of a prospective study of combined modality therapy for stage I-II indolent non-Hodgkin's lymphoma. J Clin Oncol 21 (11): 2115-22, 2003.
- Advani R, Rosenberg SA, Horning SJ: Stage I and II follicular non-Hodgkin's lymphoma: long-term follow-up of no initial therapy. J Clin Oncol 22 (8): 1454-9, 2004.
- Pugh TJ, Ballonoff A, Newman F, et al.: Improved survival in patients with early stage low-grade follicular lymphoma treated with radiation: a Surveillance, Epidemiology, and End Results database analysis. Cancer 116 (16): 3843-51, 2010.
Treatment of Indolent, Noncontiguous Stage II / III / IV NHL
Optimal treatment of advanced stages of low-grade non-Hodgkin lymphoma (NHL) is controversial because of low cure rates with the current therapeutic options. Numerous clinical trials are in progress to evaluate treatment issues, and patients are urged to participate. The rate of relapse is fairly constant over time, even in patients who have achieved complete response to treatment. Indeed, relapse may occur many years after treatment. Currently, no randomized trials provide guidance to clinicians about the initial choice of watchful waiting, rituximab, nucleoside analogues, alkylating agents, combination chemotherapy, radiolabeled monoclonal antibodies, or combinations of these options.[1]; [2][Level of evidence B1]
For patients with indolent, noncontiguous stage II and stage III NHL, central lymphatic radiation therapy has been proposed but is not usually recommended as a form of treatment.[3,4]
Patients with a resolved hepatitis B virus (HBV) infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. Prophylactic nucleoside therapy lowered HBV reactivation from 10.8% to 2.1% in a retrospective study of 326 patients.[5]
Treatment Options for Indolent, Noncontiguous Stage II/III/IV NHL
Treatment options for indolent, noncontiguous stage II/III/IV NHL include the following:
- Watchful waiting for asymptomatic patients.
- Rituximab alone or in combination with cytotoxic agents used in front-line therapy.
- Lenalidomide and rituximab.
- Maintenance rituximab.
- Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy.
- Phosphatidylinositol 3-kinase (PI3K) inhibitor.
- EZH2 inhibitor.
- Radiolabeled anti-CD20 monoclonal antibodies.
- Intensive therapy with chemotherapy with or without total-body irradiation or high-dose radioimmunotherapy followed by autologous or allogeneic bone marrow transplantation or peripheral stem cell transplantation (under clinical evaluation).[6,7,8,9,10,11,12,13,14,15]
- Phase III clinical trials comparing chemotherapy alone versus chemotherapy followed by anti-idiotype vaccine.[16,17,18]
- Extended-field radiation therapy (stage III patients only) (under clinical evaluation).[19]
- Ofatumumab—human anti–CD20 monoclonal antibody (under clinical evaluation).[20]
- Short-course low-dose, palliative radiation therapy (2 × 2 Gy) (under clinical evaluation).[21,22]
Because none of the therapies listed above are curative for advanced-stage disease, innovative approaches are under clinical evaluation.
Watchful waiting for asymptomatic patients
The rate of relapse is fairly constant over time, even in patients who have achieved complete responses (CR) to treatment. Indeed, relapse may occur many years after treatment. In this category, deferred treatment (i.e., watchful waiting until the patient becomes symptomatic before initiating treatment) can be considered.[2,23,24,25] The Follicular Lymphoma International Prognostic Index (FLIPI) and the revised FLIPI-2 can predict progression-free survival (PFS) and overall survival (OS), but the scores cannot be used to establish the need for therapy in asymptomatic patients.[26,27]
Evidence (watchful waiting):
- Three randomized trials compared watchful waiting with immediate chemotherapy.[24,28]; [29][Level of evidence A1]
- All three trials showed no difference in cause-specific or OS.
- For patients randomly assigned to watchful waiting, the median time to require therapy was 2 to 3 years and one-third of patients receiving watchful waiting never required treatment with watchful waiting (half died of other causes and half remained progression free after 10 years).
- A selected group of 107 patients with advanced-stage follicular lymphoma were managed with initial watchful waiting; with a median delay of 55 months, subsequent therapy resulted in equivalent freedom from treatment failure and OS compared with a similar cohort treated immediately with rituximab.[30][Level of evidence C2] This implies that watchful waiting remains a relevant approach even in the rituximab era.
Rituximab alone or in combination with cytotoxic agents used in front-line therapy
Standard therapy includes rituximab, an anti–CD20 monoclonal antibody, either alone, as was shown in the ECOG-E4402 trial (NCT00075946),[31,32,33,34,35] or in combination with purine nucleoside analogues, such as fludarabine or cladribine, alkylating agents (with or without steroids), or combination chemotherapy. Rituximab may be considered as first-line therapy, either alone or in combination with other agents. Rituximab may be given intravenously or subcutaneously, and biosimilar versions, such as CT-P10 and GP2013, have shown equivalent efficacy and safety.[36,37,38] Combinations include the following:
- R-bendamustine: rituximab + bendamustine.[39,40,41]
- R-F: rituximab + fludarabine.[42]
- R-CVP: rituximab + cyclophosphamide + vincristine + prednisone.[43,44,45,46]
- R-CHOP: rituximab + cyclophosphamide + doxorubicin + vincristine + prednisone.[45,46,47,48,49] A Cochrane meta-analysis could not identify any OS benefit of adding doxorubicin to chemotherapy regimens with rituximab or to chemotherapy regimens without rituximab.[50][Level of evidence A1]
- R-FM: rituximab + fludarabine + mitoxantrone.[45,46,51]
- R-FCM: rituximab + fludarabine + cyclophosphamide + mitoxantrone.[52]
Evidence (rituximab with or without chemotherapy):
- A prospective, randomized trial of 534 patients with previously untreated, advanced-stage follicular lymphoma compared R-CHOP, R-FM, and R-CVP.[45]
- With a median follow-up of 84 months, there was no difference in OS (8-year OS rate, 83%; 95% confidence interval (CI), 79%–87%), but the 8-year PFS rates favored R-CHOP (52%) and R-FM (49%) over R-CVP (42%) (P for the three regimens = .037).[45][Level of evidence B1]
- Four randomized prospective studies of previously untreated patients (involving more than 1,300 patients) and one Cochrane meta-analysis that included both untreated and previously treated patients (involving almost 1,000 patients) have compared rituximab plus combination chemotherapy with chemotherapy alone.[44,49,53]; [54,55][Level of evidence A1]
- Rituximab plus chemotherapy was superior in terms of event-free survival (EFS) or PFS (ranging from 2–3 years) in all of the studies and in terms of OS in all but one study (absolute benefit ranging from 6%–13% at 4 years, P < .04 and hazard ratio [HR] = 0.63 [0.51–0.79] for the meta-analysis).
- All of these trials were performed in symptomatic patients who required therapy. These results do not negate watchful waiting when appropriate.
- Fluorine F 18-fludeoxyglucose positron emission tomography–computed tomography (18F-FDG PET-CT) scan status at the completion of rituximab plus chemotherapy induction therapy is strongly predictive of outcome. It is not yet known whether acting on the results of the scans translates into better outcomes.[56,57]
- In a prospective randomized trial (NCT00991211), 527 patients with indolent and mantle cell lymphoma were randomly assigned to a bendamustine-and-rituximab arm versus an R-CHOP arm.[40][Level of evidence B1]
- With a median follow-up of 45 months, the median PFS favored the bendamustine arm (69 months vs. 31 months [HR, 0.58; 95% CI, 0.44–0.74; P < .0001]) but with no difference in OS.
- The bendamustine arm was associated with significantly lower rates of alopecia, hematologic toxicity, stomatitis, peripheral neuropathy, and infections than was the R-CHOP arm.
- In a similar prospective randomized trial, 447 patients with indolent and mantle cell lymphoma were assigned to bendamustine and rituximab versus R-CHOP or R-CVP.[41][Level of evidence B1]
- With a median follow-up of 65 months, the 5-year PFS rate favored bendamustine and rituximab, 65.5% versus 55.8% (HR, 0.61; 95% CI, 0.45–0.85; P = .0025), but with no difference in OS.
- Increased deaths in the bendamustine-and-rituximab arm from cardiovascular causes (seven versus one) and from secondary malignancies other than lymphoma (five versus three) may have contributed to the lack of OS advantage.
Lenalidomide and rituximab
The combination of the immunomodulating agent lenalidomide with rituximab (the so-called R2 regimen) has been proposed as an alternative regimen to combinations involving cytotoxic agents and their subsequent short- and long-term toxicities.
Evidence (lenalidomide and rituximab):
- In a randomized prospective trial (RELEVANCE) of 1,030 patients with previously untreated follicular lymphoma, rituximab plus lenalidomide for 18 months was compared with rituximab plus chemotherapy (usually R-CHOP).[58,59] All patients received rituximab maintenance for up to 2 years.
- With a median follow-up of 72 months, the 6-year PFS (60% and 59%) and 3-year OS (89%) were identical (HR, 1.03; 95% CI, 0.84–1.27; P = .78 for PFS) (HR for OS was not reported).[58,59][Level of evidence A1]
- This trial established that the R2 regimen is as effective as rituximab plus cytotoxic chemotherapy options. The transformation rate to aggressive lymphoma per year was 0.68% in the R2 group and 0.45% in the R-chemotherapy group. With a median follow-up of 72 months, there were no new safety signals.[59]
- In a randomized prospective trial of 358 patients with resistant/refractory indolent lymphoma (usually follicular lymphoma), the R2 regimen was compared with rituximab alone.[60]
- With a median follow-up of 28 months, the median PFS was 39.4 months for R2 and 14.1 months for rituximab alone (P < .0001), with no difference in OS.[60][Level of evidence B1]
Maintenance rituximab
After induction therapy with rituximab only or with rituximab plus chemotherapy, rituximab can be used once every 2 to 3 months. Several studies have evaluated this approach.
Evidence (maintenance rituximab for previously untreated patients):
- In the PRIMA study (NCT00140582), 1,018 high-risk, previously untreated, symptomatic patients achieved CR or partial response (PR) after induction therapy with immunochemotherapy (usually R-CHOP) and were then randomly assigned to 2 years of maintenance rituximab versus no maintenance.[61][Level of evidence B1]
- With a median follow-up of 9.0 years, median PFS favored rituximab maintenance (10.5 years) compared with observation (4.1 years) (HR, 0.61; 95% CI, 0.52−0.73; P < .001), but with no difference in OS.
- In the United Kingdom/International Study (NCT00112931), 379 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to watchful waiting versus rituximab induction only versus rituximab induction followed by 2 years of rituximab maintenance.[62][Level of evidence A3]
- Although OS and histological transformation rates were no different at 3 years, maintenance rituximab was favored based on quality-of-life (QOL) studies (Mental Adjustment to Cancer Scale P = .0004 at 7 months; Illness Coping Score P = .0012 at 7 months) and time-to-initiation of new treatment by 3 years (54% for watchful waiting vs. 12% for rituximab maintenance [HR, 0.21; 95% CI, 0.14–0.31; P < .0001]).[62][Level of evidence A3]
- This study suggested that for some patients, watch and wait resulted in watch and worry.[63] However, from the perspective of OS and histological transformation rates, no benefit could be seen with rituximab maintenance.
- In the RESORT study (NCT00075946), 289 previously untreated patients with asymptomatic, low-burden disease were randomly assigned to rituximab induction alone, with a re-treatment strategy that used rituximab at relapse, or rituximab induction plus maintenance rituximab every 13 weeks until treatment failure.[34][Level of evidence A3]
- With a median follow-up of 4.5 years, there was no difference in median time-to-treatment failure (defined as failing rituximab alone) or in health-related QOL. A re-treatment strategy achieved comparable disease control using significantly fewer doses of rituximab.
These three randomized trials in previously untreated patients show no advantage for the use of rituximab maintenance versus observation and reinduction of therapy at the time of relapse. The trials suggest a benefit for maintenance rituximab after reinduction for relapsed disease. Many questions remain about rituximab maintenance, particularly about truncating therapy at 2 years and long-term safety and efficacy. A trial extending rituximab maintenance to 5 years showed similar EFS or OS versus 1 year of maintenance after induction therapy with rituximab in previously untreated patients.[64][Level of evidence A1]
- The FOLL12 study (NCT02063685) included 807 patients with previously untreated high-tumor burden follicular lymphoma. Patients received rituximab plus chemotherapy induction and were randomly assigned to either standard rituximab maintenance (every 8 weeks for 2 years) or to postinduction treatment (monitoring, rituximab maintenance, or radioimmunotherapy) based on their complete metabolic response and measurable residual disease–negative status.[65]
- With a median follow-up of 53 months, the 3-year PFS rate was 86% for patients who received standard maintenance and 72% for patients who received response-based treatment (P < .001). The 3-year OS was the same in both groups (98% vs. 97%; P = .238).[65][Level of evidence B1]
- This trial does not support the use of an end-of-treatment PET-CT scan to guide the use of maintenance rituximab.
For previously untreated patients, all of the studies showed improvement of PFS, with no change in OS.
Evidence (maintenance rituximab for previously treated patients):
- In a prospective randomized trial of 465 patients with relapsed follicular lymphoma, responders to R-CHOP or CHOP were further randomly assigned to receive rituximab maintenance (1 dose every 3 months for 2 years) or no maintenance.[66][Level of evidence B1]
- At a median follow-up of 6 years, rituximab maintenance was better for median PFS (44 months vs. 16 months, P < .001) and borderline for 5-year OS (74% vs. 64%, P = .07).
- This benefit for maintenance was evident even for patients who received rituximab during induction therapy. Most patients in both arms received extensive rituximab during postprotocol salvage treatment.
- In a prospective randomized trial of 280 patients with relapsed follicular lymphoma, responders to chemotherapy and autologous stem cell transplantation consolidation were randomly assigned to receive four doses of rituximab maintenance or no maintenance.[67][Level of evidence B1]
- With an 8.3-year median follow-up, the 10-year PFS rates favored maintenance (54% vs. 37% [HR, 0.66; 95% CI, 0.47–0.91; P = .012]), but there was no difference in OS.
- A meta-analysis of nine randomized clinical trials with a total of 2,586 patients with follicular lymphoma, most of whom had relapsed disease, compared rituximab maintenance with no maintenance and showed improved OS for rituximab maintenance in previously treated patients (HRdeath, 0.72; 95% CI, 0.57–0.91).[68][Level of evidence A1]
For previously treated patients, there is more evidence to suggest an OS advantage with the use of rituximab maintenance.
Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy
Obinutuzumab is a glycoengineered type II anti–CD20 monoclonal antibody with greater antibody-dependent cellular cytotoxicity than rituximab.
Evidence (obinutuzumab):
- A prospective randomized trial (NCT01332968) of 1,202 patients with previously untreated follicular lymphoma compared obinutuzumab combined with bendamustine (50%), cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) (33%), or cyclophosphamide, vincristine, and prednisone (CVP) (10%) with rituximab combined with the same chemotherapy regimens (based on investigator choice).[69] After six cycles of combination therapy, patients had 2 years of maintenance therapy, receiving the same antibody every 2 months.
- With a median follow-up of 34.5 months, the 3-year PFS rate was 80% in the obinutuzumab group and 73.3% in the rituximab group (HR, 0.66; 95% CI, 0.51–0.85, P = .001).[69][Level of evidence B1]
- There was no difference in OS.
- There was a high rate of toxic deaths among patients using bendamustine in the obinutuzumab arm (5.6%) and in the rituximab arm (4.4%) compared with what has been seen historically. For patients with indolent low-grade lymphoma, with median survivals exceeding 15 years, the number of toxic deaths during first-line therapy seems excessive. By comparison, the toxic death rate was 1% to 2% when either antibody was combined with CHOP or CVP.
Several issues have been raised about this study:
- The side effects were significantly higher with obinutuzumab in terms of infusion reactions and subsequent adverse events.
- Obinutuzumab costs significantly more than rituximab.
In summary, in the absence of any change in OS, switching from rituximab to obinutuzumab in combination with chemotherapy for previously untreated follicular lymphoma is a difficult choice. The PFS differences may be attributable to the imbalance in monoclonal antibody dosing, and the increased side effects and costs are mitigating factors. In this trial, bendamustine combined with either antibody led to unacceptable rates of toxic death.
Phosphatidylinositol 3-kinase (PI3K) inhibitor
Copanlisib
Evidence (copanlisib):
- A double-blind, randomized, placebo-controlled study assigned 307 patients with recurrent indolent lymphoma to copanlisib plus rituximab and 151 patients to placebo plus rituximab (2:1 randomization).[70]
- With a median follow-up of 19.2 months, the median PFS was 21.5 months for patients in the copanlisib-plus-rituximab arm (95% CI, 17.8–33.0) versus 13.8 months (95% CI, 10.2–17.5) for patients in the placebo-plus-rituximab arm (HR, 0.52; 95% CI, 0.39–0.69; P < .0001).[70][Level of evidence B1]
- Hyperglycemia and hypertension were the most common grade 3 to 4 adverse events, occurring in half of the patients who received copanlisib.
The PI3K inhibitors have significant adverse effects, including pneumonitis, colitis, transaminitis, hypertension, hyperglycemia, rash, and increased risk of infections. These adverse events have affected the use of these agents until confirmatory trial results can establish their efficacy and safety.[71] They are currently approved for treatment of relapsed and refractory follicular lymphoma after two previously received lines of therapy.
EZH2 inhibitor
Tazemetostat
Tazemetostat is an inhibitor of EZH2, a histone methyltransferase essential to the formation of lymph node germinal centers, especially with activating mutations of EZH2.
Evidence (tazemetostat):
- A phase II study included 99 patients with relapsed or refractory follicular lymphoma, 45 of whom had activating mutations of EZH2, and 54 of whom had normal wild-type EZH2.[72]
- Treatment with tazemetostat resulted in an objective response rate of 69% (95% CI, 53%–82%) for patients with activating mutations versus 35% (95% CI, 23%–49%) for patients with wild-type EZH2.[72]
- With a median follow-up of 22 months, the median PFS was 13.8 months (95% CI, 10.7–22.0) for patients with activating mutations and 11.1 months (95% CI, 3.7–14.6) for patients with wild-type EZH2.[72][Level of evidence C3]
- Grade 3 or 4 treatment-related adverse events were seen in 4% of patients.
Radiolabeled anti-CD20 monoclonal antibodies
Yttrium Y 90 (90Y)-ibritumomab tiuxetan (Zevalin) is available for previously untreated and relapsing patients with minimal (<25%) or no marrow involvement with lymphoma (iodine I 131 [131I]-tositumomab [Bexxar] is no longer available because of commercial disengagement).[73,74]
Evidence (radiolabeled anti-CD20 monoclonal antibodies):
- In a randomized, prospective trial, 554 patients with previously untreated advanced-stage follicular lymphoma received either R-CHOP times six cycles or CHOP times six cycles followed by 131I-tositumomab radioimmunotherapy (RIT).[75]
- With a median follow-up of 10.3 years, there was improvement in PFS for RIT (56% vs. 42%, P = .01) but no significant difference in 10-year OS (OS R-CHOP, 81%; CHOP-RIT, 75%; P = 0.13).[75][Level of evidence B1]
- In this trial, the cumulative incidence of death from myelodysplastic syndrome or acute myeloid leukemia is higher for the RIT arm (4% vs. 1%, P = .02).
131I-tositumomab became commercially unavailable in 2013.
- In a randomized trial of 409 patients with stage III or IV follicular lymphoma who achieved a CR or PR, 90Y-ibritumomab tiuxetan consolidation versus no consolidation was evaluated.[76]
- The radiolabeled antibody consolidation improved median PFS by 3 years (P < .001), and median time to next treatment was improved by 5.1 years (P < .001); however, there was no change in OS.[76][Level of evidence B1]
Durable responses to radiolabeled monoclonal antibodies, such as 90Y-ibritumomab tiuxetan (commercially available) and iodine I 131-tositumomab (commercially unavailable), have also been reported before and after cytotoxic chemotherapy.[75,77,78][Level of evidence B1] However, the cumulative incidence of death resulting from myelodysplastic syndrome or acute myeloid leukemia is higher (4% vs. 1%; P = .02) in one of the randomized trials versus nonradiolabeled antibody with chemotherapy.[75]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Hagenbeek A, Eghbali H, Monfardini S, et al.: Phase III intergroup study of fludarabine phosphate compared with cyclophosphamide, vincristine, and prednisone chemotherapy in newly diagnosed patients with stage III and IV low-grade malignant Non-Hodgkin's lymphoma. J Clin Oncol 24 (10): 1590-6, 2006.
- Gribben JG: How I treat indolent lymphoma. Blood 109 (11): 4617-26, 2007.
- Jacobs JP, Murray KJ, Schultz CJ, et al.: Central lymphatic irradiation for stage III nodular malignant lymphoma: long-term results. J Clin Oncol 11 (2): 233-8, 1993.
- Mendenhall NP, Million RR: Comprehensive lymphatic irradiation for stage II-III non-Hodgkin's lymphoma. Am J Clin Oncol 12 (3): 190-4, 1989.
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- van Besien K, Sobocinski KA, Rowlings PA, et al.: Allogeneic bone marrow transplantation for low-grade lymphoma. Blood 92 (5): 1832-6, 1998.
- van Besien K, Loberiza FR, Bajorunaite R, et al.: Comparison of autologous and allogeneic hematopoietic stem cell transplantation for follicular lymphoma. Blood 102 (10): 3521-9, 2003.
- Deconinck E, Foussard C, Milpied N, et al.: High-dose therapy followed by autologous purged stem-cell transplantation and doxorubicin-based chemotherapy in patients with advanced follicular lymphoma: a randomized multicenter study by GOELAMS. Blood 105 (10): 3817-23, 2005.
- Sebban C, Mounier N, Brousse N, et al.: Standard chemotherapy with interferon compared with CHOP followed by high-dose therapy with autologous stem cell transplantation in untreated patients with advanced follicular lymphoma: the GELF-94 randomized study from the Groupe d'Etude des Lymphomes de l'Adulte (GELA). Blood 108 (8): 2540-4, 2006.
- Lenz G, Dreyling M, Schiegnitz E, et al.: Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 104 (9): 2667-74, 2004.
- Rohatiner AZ, Nadler L, Davies AJ, et al.: Myeloablative therapy with autologous bone marrow transplantation for follicular lymphoma at the time of second or subsequent remission: long-term follow-up. J Clin Oncol 25 (18): 2554-9, 2007.
- Gopal AK, Rajendran JG, Gooley TA, et al.: High-dose [131I]tositumomab (anti-CD20) radioimmunotherapy and autologous hematopoietic stem-cell transplantation for adults > or = 60 years old with relapsed or refractory B-cell lymphoma. J Clin Oncol 25 (11): 1396-402, 2007.
- Gyan E, Foussard C, Bertrand P, et al.: High-dose therapy followed by autologous purged stem cell transplantation and doxorubicin-based chemotherapy in patients with advanced follicular lymphoma: a randomized multicenter study by the GOELAMS with final results after a median follow-up of 9 years. Blood 113 (5): 995-1001, 2009.
- Al Khabori M, de Almeida JR, Guyatt GH, et al.: Autologous stem cell transplantation in follicular lymphoma: a systematic review and meta-analysis. J Natl Cancer Inst 104 (1): 18-28, 2012.
- Schaaf M, Reiser M, Borchmann P, et al.: High-dose therapy with autologous stem cell transplantation versus chemotherapy or immuno-chemotherapy for follicular lymphoma in adults. Cochrane Database Syst Rev 1: CD007678, 2012.
- Bendandi M, Gocke CD, Kobrin CB, et al.: Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat Med 5 (10): 1171-7, 1999.
- Neelapu SS, Gause BL, Nikcevich DA, et al.: Phase III randomized trial of patient-specific vaccination for previously untreated patients with follicular lymphoma in first complete remission: protocol summary and interim report. Clin Lymphoma 6 (1): 61-4, 2005.
- Inogès S, Rodrìguez-Calvillo M, Zabalegui N, et al.: Clinical benefit associated with idiotypic vaccination in patients with follicular lymphoma. J Natl Cancer Inst 98 (18): 1292-301, 2006.
- Ha CS, Kong JS, Tucker SL, et al.: Central lymphatic irradiation for stage I-III follicular lymphoma: report from a single-institutional prospective study. Int J Radiat Oncol Biol Phys 57 (2): 316-20, 2003.
- Czuczman MS, Fayad L, Delwail V, et al.: Ofatumumab monotherapy in rituximab-refractory follicular lymphoma: results from a multicenter study. Blood 119 (16): 3698-704, 2012.
- Chan EK, Fung S, Gospodarowicz M, et al.: Palliation by low-dose local radiation therapy for indolent non-Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 81 (5): e781-6, 2011.
- Rossier C, Schick U, Miralbell R, et al.: Low-dose radiotherapy in indolent lymphoma. Int J Radiat Oncol Biol Phys 81 (3): e1-6, 2011.
- Eek R, Falkson G: The low-grade lymphoproliferative disorders. Oncology 54 (6): 441-58, 1997 Nov-Dec.
- Ardeshna KM, Smith P, Norton A, et al.: Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet 362 (9383): 516-22, 2003.
- Portlock CS, Rosenberg SA: No initial therapy for stage III and IV non-Hodgkin's lymphomas of favorable histologic types. Ann Intern Med 90 (1): 10-13, 1979.
- Solal-Céligny P, Roy P, Colombat P, et al.: Follicular lymphoma international prognostic index. Blood 104 (5): 1258-65, 2004.
- Federico M, Bellei M, Marcheselli L, et al.: Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol 27 (27): 4555-62, 2009.
- Brice P, Bastion Y, Lepage E, et al.: Comparison in low-tumor-burden follicular lymphomas between an initial no-treatment policy, prednimustine, or interferon alfa: a randomized study from the Groupe d'Etude des Lymphomes Folliculaires. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1110-7, 1997.
- Young RC, Longo DL, Glatstein E, et al.: The treatment of indolent lymphomas: watchful waiting v aggressive combined modality treatment. Semin Hematol 25 (2 Suppl 2): 11-6, 1988.
- Solal-Céligny P, Bellei M, Marcheselli L, et al.: Watchful waiting in low-tumor burden follicular lymphoma in the rituximab era: results of an F2-study database. J Clin Oncol 30 (31): 3848-53, 2012.
- Ghielmini M, Schmitz SF, Cogliatti SB, et al.: Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly x 4 schedule. Blood 103 (12): 4416-23, 2004.
- Witzig TE, Vukov AM, Habermann TM, et al.: Rituximab therapy for patients with newly diagnosed, advanced-stage, follicular grade I non-Hodgkin's lymphoma: a phase II trial in the North Central Cancer Treatment Group. J Clin Oncol 23 (6): 1103-8, 2005.
- Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin's lymphoma--a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005.
- Kahl BS, Hong F, Williams ME, et al.: Rituximab extended schedule or re-treatment trial for low-tumor burden follicular lymphoma: eastern cooperative oncology group protocol e4402. J Clin Oncol 32 (28): 3096-102, 2014.
- Buske C, Hiddemann W: Rituximab maintenance therapy in indolent NHL: a clinical review. Leuk Res 30 (Suppl 1): S11-5, 2006.
- Kim WS, Buske C, Ogura M, et al.: Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol 4 (8): e362-e373, 2017.
- Davies A, Merli F, Mihaljević B, et al.: Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol 4 (6): e272-e282, 2017.
- Jurczak W, Moreira I, Kanakasetty GB, et al.: Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol 4 (8): e350-e361, 2017.
- Robinson KS, Williams ME, van der Jagt RH, et al.: Phase II multicenter study of bendamustine plus rituximab in patients with relapsed indolent B-cell and mantle cell non-Hodgkin's lymphoma. J Clin Oncol 26 (27): 4473-9, 2008.
- Rummel MJ, Niederle N, Maschmeyer G, et al.: Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381 (9873): 1203-10, 2013.
- Flinn IW, van der Jagt R, Kahl B, et al.: First-Line Treatment of Patients With Indolent Non-Hodgkin Lymphoma or Mantle-Cell Lymphoma With Bendamustine Plus Rituximab Versus R-CHOP or R-CVP: Results of the BRIGHT 5-Year Follow-Up Study. J Clin Oncol 37 (12): 984-991, 2019.
- Czuczman MS, Koryzna A, Mohr A, et al.: Rituximab in combination with fludarabine chemotherapy in low-grade or follicular lymphoma. J Clin Oncol 23 (4): 694-704, 2005.
- Marcus R, Imrie K, Belch A, et al.: CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 105 (4): 1417-23, 2005.
- Marcus R, Imrie K, Solal-Celigny P, et al.: Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol 26 (28): 4579-86, 2008.
- Federico M, Luminari S, Dondi A, et al.: R-CVP versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage follicular lymphoma: results of the FOLL05 trial conducted by the Fondazione Italiana Linfomi. J Clin Oncol 31 (12): 1506-13, 2013.
- Luminari S, Ferrari A, Manni M, et al.: Long-Term Results of the FOLL05 Trial Comparing R-CVP Versus R-CHOP Versus R-FM for the Initial Treatment of Patients With Advanced-Stage Symptomatic Follicular Lymphoma. J Clin Oncol 36 (7): 689-696, 2018.
- Czuczman MS, Weaver R, Alkuzweny B, et al.: Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin's lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J Clin Oncol 22 (23): 4711-6, 2004.
- Hainsworth JD, Litchy S, Morrissey LH, et al.: Rituximab plus short-duration chemotherapy as first-line treatment for follicular non-Hodgkin's lymphoma: a phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (7): 1500-6, 2005.
- Hiddemann W, Kneba M, Dreyling M, et al.: Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 106 (12): 3725-32, 2005.
- Itchaki G, Gafter-Gvili A, Lahav M, et al.: Anthracycline-containing regimens for treatment of follicular lymphoma in adults. Cochrane Database Syst Rev 7: CD008909, 2013.
- Zinzani PL, Pulsoni A, Perrotti A, et al.: Fludarabine plus mitoxantrone with and without rituximab versus CHOP with and without rituximab as front-line treatment for patients with follicular lymphoma. J Clin Oncol 22 (13): 2654-61, 2004.
- Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004.
- Herold M, Haas A, Srock S, et al.: Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol 25 (15): 1986-92, 2007.
- Salles GA, Mounier N, de Guibert S, et al.: Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: final analysis of the GELA-GOELAMS FL2000 study with a 5-year follow-up. [Abstract] Blood 110 (11): A-792, 2007.
- Schulz H, Bohlius J, Skoetz N, et al.: Combined immunochemotherapy with rituximab improves overall survival in patients with follicular and mantle cell lymphoma: updated meta-analysis results. [Abstract] Blood 108 (11): A-2760, 2006.
- Dupuis J, Berriolo-Riedinger A, Julian A, et al.: Impact of [(18)F]fluorodeoxyglucose positron emission tomography response evaluation in patients with high-tumor burden follicular lymphoma treated with immunochemotherapy: a prospective study from the Groupe d'Etudes des Lymphomes de l'Adulte and GOELAMS. J Clin Oncol 30 (35): 4317-22, 2012.
- Trotman J, Fournier M, Lamy T, et al.: Positron emission tomography-computed tomography (PET-CT) after induction therapy is highly predictive of patient outcome in follicular lymphoma: analysis of PET-CT in a subset of PRIMA trial participants. J Clin Oncol 29 (23): 3194-200, 2011.
- Morschhauser F, Fowler NH, Feugier P, et al.: Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med 379 (10): 934-947, 2018.
- Morschhauser F, Nastoupil L, Feugier P, et al.: Six-Year Results From RELEVANCE: Lenalidomide Plus Rituximab (R2) Versus Rituximab-Chemotherapy Followed by Rituximab Maintenance in Untreated Advanced Follicular Lymphoma. J Clin Oncol 40 (28): 3239-3245, 2022.
- Leonard JP, Trneny M, Izutsu K, et al.: AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37 (14): 1188-1199, 2019.
- Bachy E, Seymour JF, Feugier P, et al.: Sustained Progression-Free Survival Benefit of Rituximab Maintenance in Patients With Follicular Lymphoma: Long-Term Results of the PRIMA Study. J Clin Oncol 37 (31): 2815-2824, 2019.
- Ardeshna KM, Qian W, Smith P, et al.: Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: an open-label randomised phase 3 trial. Lancet Oncol 15 (4): 424-35, 2014.
- Ansell SM: Follicular lymphoma: watch and wait is watch and worry. Lancet Oncol 15 (4): 368-9, 2014.
- Taverna C, Martinelli G, Hitz F, et al.: Rituximab Maintenance for a Maximum of 5 Years After Single-Agent Rituximab Induction in Follicular Lymphoma: Results of the Randomized Controlled Phase III Trial SAKK 35/03. J Clin Oncol 34 (5): 495-500, 2016.
- Luminari S, Manni M, Galimberti S, et al.: Response-Adapted Postinduction Strategy in Patients With Advanced-Stage Follicular Lymphoma: The FOLL12 Study. J Clin Oncol 40 (7): 729-739, 2022.
- van Oers MH, Tönnissen E, Van Glabbeke M, et al.: BCL-2/IgH polymerase chain reaction status at the end of induction treatment is not predictive for progression-free survival in relapsed/resistant follicular lymphoma: results of a prospective randomized EORTC 20981 phase III intergroup study. J Clin Oncol 28 (13): 2246-52, 2010.
- Pettengell R, Schmitz N, Gisselbrecht C, et al.: Rituximab purging and/or maintenance in patients undergoing autologous transplantation for relapsed follicular lymphoma: a prospective randomized trial from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 31 (13): 1624-30, 2013.
- Vidal L, Gafter-Gvili A, Salles G, et al.: Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst 103 (23): 1799-806, 2011.
- Marcus R, Davies A, Ando K, et al.: Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med 377 (14): 1331-1344, 2017.
- Matasar MJ, Capra M, Özcan M, et al.: Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 22 (5): 678-689, 2021.
- Richardson NC, Kasamon Y, Pazdur R, et al.: The saga of PI3K inhibitors in haematological malignancies: survival is the ultimate safety endpoint. Lancet Oncol 23 (5): 563-566, 2022.
- Morschhauser F, Tilly H, Chaidos A, et al.: Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21 (11): 1433-1442, 2020.
- Press OW, Unger JM, Braziel RM, et al.: Phase II trial of CHOP chemotherapy followed by tositumomab/iodine I-131 tositumomab for previously untreated follicular non-Hodgkin's lymphoma: five-year follow-up of Southwest Oncology Group Protocol S9911. J Clin Oncol 24 (25): 4143-9, 2006.
- Scholz CW, Pinto A, Linkesch W, et al.: (90)Yttrium-ibritumomab-tiuxetan as first-line treatment for follicular lymphoma: 30 months of follow-up data from an international multicenter phase II clinical trial. J Clin Oncol 31 (3): 308-13, 2013.
- Shadman M, Li H, Rimsza L, et al.: Continued Excellent Outcomes in Previously Untreated Patients With Follicular Lymphoma After Treatment With CHOP Plus Rituximab or CHOP Plus 131I-Tositumomab: Long-Term Follow-Up of Phase III Randomized Study SWOG-S0016. J Clin Oncol 36 (7): 697-703, 2018.
- Morschhauser F, Radford J, Van Hoof A, et al.: 90Yttrium-ibritumomab tiuxetan consolidation of first remission in advanced-stage follicular non-Hodgkin lymphoma: updated results after a median follow-up of 7.3 years from the International, Randomized, Phase III First-LineIndolent trial. J Clin Oncol 31 (16): 1977-83, 2013.
- Fisher RI, Kaminski MS, Wahl RL, et al.: Tositumomab and iodine-131 tositumomab produces durable complete remissions in a subset of heavily pretreated patients with low-grade and transformed non-Hodgkin's lymphomas. J Clin Oncol 23 (30): 7565-73, 2005.
- Leahy MF, Seymour JF, Hicks RJ, et al.: Multicenter phase II clinical study of iodine-131-rituximab radioimmunotherapy in relapsed or refractory indolent non-Hodgkin's lymphoma. J Clin Oncol 24 (27): 4418-25, 2006.
Treatment of Indolent, Recurrent NHL
In general, treatment with standard agents rarely produces a cure in patients whose disease has relapsed. Sustained remissions after relapse can often be obtained in patients with indolent lymphomas, but relapse will usually ensue. Favorable survival after relapse has been associated with an age younger than 60 years, complete remission rather than partial remission, and duration of response longer than 2 years.[1] Even the most favorable subset, however, has a tenfold greater mortality compared with age-adjusted U.S. population rates.[2]
Patients who experience a relapse with indolent lymphoma can often have their disease controlled with single agent or combination chemotherapy, rituximab (an anti–CD20 monoclonal antibody), lenalidomide, radiolabeled anti–CD20 monoclonal antibodies, or palliative radiation therapy.[3,4] Long-term freedom from second relapse, however, is uncommon and multiple relapses will usually occur. Patients with indolent lymphoma may experience a relapse with a more aggressive histology. If the clinical pattern of relapse suggests that the disease is behaving in a more aggressive manner, a biopsy can be performed. Documentation of conversion to a more aggressive histology requires an appropriate change to therapy applicable to that histological type.[5] Rapid growth or discordant growth between various disease sites may indicate a histological conversion.
In a retrospective review of 325 patients between 1972 and 1999, the risk of histological transformation was 30% by 10 years from diagnosis.[6] In this series, high-risk factors for subsequent histological transformation were advanced stage, high-risk Follicular Lymphoma International Prognostic Index, and expectant management. The median survival after transformation was 1 to 2 years, with 25% of patients alive at 5 years and with approximately 10% to 20% of patients alive 10 years after re-treatment.[7]
A prospective trial of 631 patients with follicular lymphoma and with a median follow-up of 60 months in the rituximab era (2002–2009) found a 5-year transformation rate (11%) to a higher-grade histology.[8] The median overall survival (OS) after transformation was 50 months, and the 5-year OS rate was 66%, if the transformation occurred more than 18 months after a diagnosis of follicular lymphoma. This series describes a better prognosis for patients with transformation than was experienced by patients in the prerituximab era.
For descriptions of the regimens used to treat histological conversions, see the Treatment of Aggressive, Recurrent NHL section. The durability of the second remission may be short, and clinical trials can be considered.
Treatment Options for Indolent, Recurrent NHL
Treatment options for indolent, recurrent non-Hodgkin lymphoma (NHL) include the following:
- Rituximab alone or in combination with cytotoxic agents used in front-line therapy.
- Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy.
- Lenalidomide and rituximab.
- Phosphatidylinositol 3-kinase (PI3K) inhibitor.
- EZH2 inhibitor.
- Palliative radiation therapy.
- Chemotherapy (single agent or combination).
- Radiolabeled anti-CD20 monoclonal antibodies.
- Chimeric antigen receptor (CAR) T-cell therapy.
- Bispecific T-cell engagers.
- Stem cell transplantation (SCT).
Rituximab alone or in combination with cytotoxic agents used in front-line therapy
Rituximab results in a 40% to 50% response rate in patients who relapse with indolent B-cell lymphomas.[9,10,11,12,13] Rituximab can also be combined with combination chemotherapy.[14,15]
Evidence (rituximab):
- In three randomized, prospective studies involving previously treated patients with relapsed indolent lymphoma, patients were randomly assigned to rituximab maintenance after re-treatment with combination chemotherapy (with or without rituximab during induction) or rituximab alone.[16,17,18]
- All trials showed prolongation of response duration,[16,17,18] and one trial demonstrated improvement in median progression-free survival (PFS) (3.7 years vs. 1.3 years, P < .001) and OS rate (74% vs. 64%, P = .07) at 5 years with a median follow-up of 39 months favoring maintenance rituximab.[17]
Obinutuzumab alone or in combination with cytotoxic agents used in front-line therapy
Obinutuzumab is a CD20-binding monoclonal antibody with alternative epitope binding.
Evidence (obinutuzumab):
- In a randomized prospective trial (NCT01059630) involving 396 patients with rituximab-refractory indolent lymphoma (mostly follicular lymphoma), patients received obinutuzumab plus bendamustine followed by obinutuzumab maintenance therapy for 2 years versus bendamustine alone with no maintenance therapy.[19,20][Level of evidence A1]
- With a median follow-up of 31.8 months, the 2-year OS rate favored the obinutuzumab combination (74.5% vs. 65.1%) (hazard ratio [HR], 0.67; 95% confidence interval [CI], 0.47–0.96; P = .027). The median PFS also favored the obinutuzumab combination (25.8 months [95% CI, 19.5–41.1 months] vs. 14.1 months [95% CI, 12.6–16.0 months]) (HR, 0.57; 95% CI, 0.44–0.73; P < .001).[20][Level of evidence A1]
- The contribution of maintenance therapy to the outcome could not be assessed in this design.
Lenalidomide and rituximab
Responses of 20% to 56% have been reported for lenalidomide, especially in patients with follicular lymphoma and small lymphocytic lymphoma, with even higher responses noted for the combination of lenalidomide and rituximab.[21,22][Level of evidence C3]
Phosphatidylinositol 3-kinase (PI3K) inhibitor
Copanlisib
Evidence (copanlisib):
- A double-blind, randomized, placebo-controlled study assigned 307 patients with recurrent indolent lymphoma to copanlisib plus rituximab and 151 patients to placebo plus rituximab (2:1 randomization).[23]
- With a median follow-up of 19.2 months, the median PFS was 21.5 months for patients in the copanlisib-plus-rituximab arm (95% CI, 17.8–33.0) versus 13.8 months (95% CI, 10.2–17.5) for patients in the placebo-plus-rituximab arm (HR, 0.52; 95% CI, 0.39–0.69; P < .0001).[23][Level of evidence B1]
- Hyperglycemia and hypertension were the most common grade 3 to 4 adverse events, occurring in half of the patients who received copanlisib.
- A phase II study included 142 patients with relapsed and refractory indolent lymphoma.[24]
- Treatment with intravenous copanlisib resulted in a 59% objective response rate (12% CR), with a median duration of response of 22.6 months.[24][Level of evidence C3]
The PI3K inhibitors have significant adverse effects, including pneumonitis, colitis, transaminitis, hypertension, hyperglycemia, rash, and increased risk of infections. These adverse events have affected the use of these agents until confirmatory trials can establish their efficacy in combinations. They are currently approved for treatment of relapsed and refractory follicular lymphoma after two previously received lines of therapy.
EZH2 inhibitor
Tazemetostat
Tazemetostat is an inhibitor of EZH2, a histone methyltransferase essential to the formation of lymph node germinal centers, especially with activating mutations of EZH2.
Evidence (tazemetostat):
- A phase II study included 99 patients with relapsed or refractory follicular lymphoma, 45 of whom had activating mutations of EZH2, and 54 of whom had normal wild-type EZH2.[25]
- Treatment with tazemetostat resulted in an objective response rate of 69% (95% CI, 53%–82%) for patients with activating mutations versus 35% (95% CI, 23%–49%) for patients with wild-type EZH2.[25]
- With a median follow-up of 22 months, the median PFS was 13.8 months (95% CI, 10.7–22.0) for patients with activating mutations and 11.1 months (95% CI, 3.7–14.6) for patients with wild-type EZH2.[25][Level of evidence C3]
- Grade 3 or 4 treatment-related adverse events were seen in 4% of patients.
Palliative radiation therapy
Palliation may be achieved with very low-dose (4 Gy) involved-field radiation therapy in two fractions for patients with indolent and aggressive relapsed disease.[26] In a prospective randomized trial, treatment with 4 Gy was inferior to treatment with 24 Gy in 12 fractions in PFS (77% vs. 92%, P < .0001).[27][Level of evidence B1]
Chemotherapy (single agent or combination)
Patients may respond to the original induction regimen again, especially if the duration of remission exceeds 1 year. For relapsing patients, rituximab alone or in combination with agents not previously used may induce remissions. For more information, see the Rituximab alone or in combination with cytotoxic agents used in front-line therapy section.
Radiolabeled anti-CD20 monoclonal antibodies
Durable responses to radiolabeled monoclonal antibodies, such as yttrium Y 90 (90Y)-ibritumomab tiuxetan (commercially available) and iodine I 131-tositumomab (commercially unavailable), have also been reported before and after cytotoxic chemotherapy.[28,29,30][Level of evidence B1] However, the cumulative incidence of death resulting from myelodysplastic syndrome or acute myeloid leukemia is higher (4% vs. 1%; P = .02) in one of the randomized trials versus nonradiolabeled antibody with chemotherapy.[30]
Evidence (radiolabeled anti-CD20 monoclonal antibodies):
- In a prospective trial of 409 patients with follicular lymphoma who responded to induction chemotherapy, patients were randomly assigned to 90Y-ibritumomab tiuxetan or no further consolidation.[31]
- With a median follow-up of 7.3 years, the 8-year PFS rate favored 90Y-ibritumomab tiuxetan (41% vs. 22% [hazard ratio, 0.47; P < .001]), but there was no difference in OS.[31][Level of evidence B1]
Chimeric antigen receptor (CAR) T-cell therapy
CAR T-cell therapy, with the autologous anti-CD19 therapeutics axicabtagene ciloleucel, lisocabtagene maraleucel, or tisagenlecleucel, has been approved for patients with relapsed follicular lymphoma after two or more lines of prior therapy.
Evidence (CAR T-cell therapy):
- In a phase II trial, 153 patients with relapsed or refractory follicular lymphoma or marginal zone lymphoma received axicabtagene ciloleucel.[32]
- With a median follow-up of 17.5 months, the overall response rate was 92% (95% CI, 85%–97%) and the CR rate was 74%.
- The 18-month PFS rate was 64.8% (95% CI, 54.2%–73.5%).[32][Level of evidence C3]
- Cytokine release syndrome occurred in 78% of patients and was grade 3 or 4 in 6% of patients.
- Tocilizumab was required in 50% of all patients, and 5% required vasopressors. Grade 3 or 4 neurological events occurred in 15% of patients.
- In a phase II trial, 98 patients with relapsed or refractory follicular lymphoma after two or more lines of prior therapy received anti-CD19 CAR T-cell therapy with tisagenlecleucel.[33]
- With a median follow-up of 16.6 months, the complete response rate was 69.1% (95% CI, 58.8%–78.3%), and the overall response rate was 86.2% (77.5%–92.4%).[33][Level of evidence C3]
- Grade 3 or 4 cytokine release syndrome occurred in 48.5% of patients, and 37.1% had grade 3 or 4 neurotoxicity.
CAR T cells are being used for high-risk patients whose disease has relapsed rapidly after chemoimmunotherapy. Such an approach is considered in the context of numerous other available agents.
Bispecific T-cell engagers
Bispecific T-cell engagers bind to CD20 (or CD19) and to CD3 to direct T cells to eliminate malignant B cells.[34,35] Similar to CAR T-cell therapy, almost one-half of patients who receive this therapy experience cytokine release syndrome.
Mosunetuzumab
Evidence (mosunetuzumab):
- In a single-arm, multicenter, phase II study, 90 patients with relapsed or refractory follicular lymphoma after two or more prior lines of therapy (including an anti-CD20 therapy and an alkylating agent) received mosunetuzumab.[34]
- With a median follow up of 27 months, the objective response rate was 77.8% (95% CI, 67.8%–85.9%), and the complete response rate was 60.0% (95% CI, 49.1%–70.2%), per investigator assessment. The median PFS per investigator assessment was not reached. The 24-month PFS was 51.4 months (95% CI, 39.4–63.3) after receiving mosunetuzumab, compared with 23.5 months (95% CI, 14.5–32.5) for patients' last prior therapy (63% chemoimmunotherapy, 8% PI3K inhibitor, 2% CAR T-cell therapy, 2% anti-CD20 antibody plus lenalidomide, and others).
- Cytokine release syndrome occurred in 44.4% of patients; 97.2% of cases were grade 1 or 2 in severity.[36][Level of evidence C3]
Stem cell transplantation
In many institutions, autologous or allogeneic SCTs are being used for high-risk patients whose disease has relapsed rapidly after chemoimmunotherapy. Such an approach is considered in the context of numerous other available agents.[37,38,39,40,41]
Evidence (SCT):
- The German Low-Grade Lymphoma Study Group treated 307 patients with follicular lymphoma with two cycles of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)-like induction chemotherapy and then randomly assigned them to autologous SCT versus interferon maintenance.[42]
- With a median follow-up of 4.2 years, the 5-year PFS rate was 65% for patients who received transplantation versus 33% for patients who received interferon (P < .001). There was no difference in OS.[42][Level of evidence B1]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Casulo C, Byrtek M, Dawson KL, et al.: Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 33 (23): 2516-22, 2015.
- Weisdorf DJ, Andersen JW, Glick JH, et al.: Survival after relapse of low-grade non-Hodgkin's lymphoma: implications for marrow transplantation. J Clin Oncol 10 (6): 942-7, 1992.
- Peterson BA: Current treatment of follicular low-grade lymphomas. Semin Oncol 26 (5 Suppl 14): 2-11, 1999.
- Haas RL, Poortmans P, de Jong D, et al.: High response rates and lasting remissions after low-dose involved field radiotherapy in indolent lymphomas. J Clin Oncol 21 (13): 2474-80, 2003.
- Tsimberidou AM, O'Brien S, Khouri I, et al.: Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24 (15): 2343-51, 2006.
- Montoto S, Davies AJ, Matthews J, et al.: Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol 25 (17): 2426-33, 2007.
- Yuen AR, Kamel OW, Halpern J, et al.: Long-term survival after histologic transformation of low-grade follicular lymphoma. J Clin Oncol 13 (7): 1726-33, 1995.
- Link BK, Maurer MJ, Nowakowski GS, et al.: Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol 31 (26): 3272-8, 2013.
- Davis TA, White CA, Grillo-López AJ, et al.: Single-agent monoclonal antibody efficacy in bulky non-Hodgkin's lymphoma: results of a phase II trial of rituximab. J Clin Oncol 17 (6): 1851-7, 1999.
- Piro LD, White CA, Grillo-López AJ, et al.: Extended Rituximab (anti-CD20 monoclonal antibody) therapy for relapsed or refractory low-grade or follicular non-Hodgkin's lymphoma. Ann Oncol 10 (6): 655-61, 1999.
- Davis TA, Grillo-López AJ, White CA, et al.: Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin's lymphoma: safety and efficacy of re-treatment. J Clin Oncol 18 (17): 3135-43, 2000.
- Hainsworth JD, Litchy S, Shaffer DW, et al.: Maximizing therapeutic benefit of rituximab: maintenance therapy versus re-treatment at progression in patients with indolent non-Hodgkin's lymphoma--a randomized phase II trial of the Minnie Pearl Cancer Research Network. J Clin Oncol 23 (6): 1088-95, 2005.
- Lockmer S, Østenstad B, Hagberg H, et al.: Chemotherapy-Free Initial Treatment of Advanced Indolent Lymphoma Has Durable Effect With Low Toxicity: Results From Two Nordic Lymphoma Group Trials With More Than 10 Years of Follow-Up. J Clin Oncol : JCO1800262, 2018.
- Forstpointner R, Dreyling M, Repp R, et al.: The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104 (10): 3064-71, 2004.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- van Oers MH, Van Glabbeke M, Giurgea L, et al.: Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin's lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 28 (17): 2853-8, 2010.
- van Oers MH, Klasa R, Marcus RE, et al.: Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non-Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 108 (10): 3295-301, 2006.
- Martinelli G, Schmitz SF, Utiger U, et al.: Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol 28 (29): 4480-4, 2010.
- Sehn LH, Chua N, Mayer J, et al.: Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol 17 (8): 1081-93, 2016.
- Cheson BD, Chua N, Mayer J, et al.: Overall Survival Benefit in Patients With Rituximab-Refractory Indolent Non-Hodgkin Lymphoma Who Received Obinutuzumab Plus Bendamustine Induction and Obinutuzumab Maintenance in the GADOLIN Study. J Clin Oncol 36 (22): 2259-2266, 2018.
- Witzig TE, Wiernik PH, Moore T, et al.: Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin's Lymphoma. J Clin Oncol 27 (32): 5404-9, 2009.
- Leonard JP, Jung SH, Johnson J, et al.: Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance). J Clin Oncol 33 (31): 3635-40, 2015.
- Matasar MJ, Capra M, Özcan M, et al.: Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 22 (5): 678-689, 2021.
- Dreyling M, Santoro A, Mollica L, et al.: Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 35 (35): 3898-3905, 2017.
- Morschhauser F, Tilly H, Chaidos A, et al.: Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21 (11): 1433-1442, 2020.
- Haas RL, Poortmans P, de Jong D, et al.: Effective palliation by low dose local radiotherapy for recurrent and/or chemotherapy refractory non-follicular lymphoma patients. Eur J Cancer 41 (12): 1724-30, 2005.
- Hoskin PJ, Kirkwood AA, Popova B, et al.: 4 Gy versus 24 Gy radiotherapy for patients with indolent lymphoma (FORT): a randomised phase 3 non-inferiority trial. Lancet Oncol 15 (4): 457-63, 2014.
- Fisher RI, Kaminski MS, Wahl RL, et al.: Tositumomab and iodine-131 tositumomab produces durable complete remissions in a subset of heavily pretreated patients with low-grade and transformed non-Hodgkin's lymphomas. J Clin Oncol 23 (30): 7565-73, 2005.
- Leahy MF, Seymour JF, Hicks RJ, et al.: Multicenter phase II clinical study of iodine-131-rituximab radioimmunotherapy in relapsed or refractory indolent non-Hodgkin's lymphoma. J Clin Oncol 24 (27): 4418-25, 2006.
- Shadman M, Li H, Rimsza L, et al.: Continued Excellent Outcomes in Previously Untreated Patients With Follicular Lymphoma After Treatment With CHOP Plus Rituximab or CHOP Plus 131I-Tositumomab: Long-Term Follow-Up of Phase III Randomized Study SWOG-S0016. J Clin Oncol 36 (7): 697-703, 2018.
- Morschhauser F, Radford J, Van Hoof A, et al.: 90Yttrium-ibritumomab tiuxetan consolidation of first remission in advanced-stage follicular non-Hodgkin lymphoma: updated results after a median follow-up of 7.3 years from the International, Randomized, Phase III First-LineIndolent trial. J Clin Oncol 31 (16): 1977-83, 2013.
- Jacobson CA, Chavez JC, Sehgal AR, et al.: Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23 (1): 91-103, 2022.
- Fowler NH, Dickinson M, Dreyling M, et al.: Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med 28 (2): 325-332, 2022.
- Budde LE, Sehn LH, Matasar M, et al.: Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol 23 (8): 1055-1065, 2022.
- Hutchings M, Mous R, Clausen MR, et al.: Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet 398 (10306): 1157-1169, 2021.
- Bartlett NL, Sehn LH, Matasar MJ, et al.: Mosunetuzumab monotherapy demonstrates durable efficacy with a manageable safety profile in patients with relapsed/refractory follicular lymphoma who received ≥2 prior therapies: updated results from a pivotal phase II study. [Abstract] Blood 140 (Suppl 1): A-610, 1467-70, 2022.
- Freedman A, Friedberg JW, Gribben J: High-dose therapy for follicular lymphoma. Oncology (Huntingt) 14 (3): 321-6, 329; discussion 330-2, 338, 2000.
- Brice P, Simon D, Bouabdallah R, et al.: High-dose therapy with autologous stem-cell transplantation (ASCT) after first progression prolonged survival of follicular lymphoma patients included in the prospective GELF 86 protocol. Ann Oncol 11 (12): 1585-90, 2000.
- Khouri IF, McLaughlin P, Saliba RM, et al.: Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood 111 (12): 5530-6, 2008.
- Sebban C, Brice P, Delarue R, et al.: Impact of rituximab and/or high-dose therapy with autotransplant at time of relapse in patients with follicular lymphoma: a GELA study. J Clin Oncol 26 (21): 3614-20, 2008.
- Thomson KJ, Morris EC, Milligan D, et al.: T-cell-depleted reduced-intensity transplantation followed by donor leukocyte infusions to promote graft-versus-lymphoma activity results in excellent long-term survival in patients with multiply relapsed follicular lymphoma. J Clin Oncol 28 (23): 3695-700, 2010.
- Lenz G, Dreyling M, Schiegnitz E, et al.: Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 104 (9): 2667-74, 2004.
Treatment of Aggressive Stage I and Aggressive, Contiguous Stage II NHL
Patients with aggressive stage I or aggressive, contiguous stage II diffuse large B-cell lymphoma (DLBCL) are candidates for combination chemotherapy with or without involved-field radiation therapy (IF-XRT).
Patients with a resolved hepatitis B virus (HBV) infection (defined as hepatitis B surface antigen-negative but hepatitis B core antibody-positive) are at risk of reactivation of HBV and require monitoring of HBV DNA. Prophylactic nucleoside therapy lowered HBV reactivation from 10.8% to 2.1% in a retrospective study of 326 patients.[1]
Treatment Options for Aggressive Stage I and Aggressive, Contiguous Stage II NHL
Treatment options for aggressive stage I and aggressive, contiguous stage II non-Hodgkin lymphoma (NHL) include the following:
- R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) with or without IF-XRT.
- R-ACVBP (rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone) (under clinical evaluation).[2,3]
R-CHOP with or without IF-XRT
The confirmation of efficacy for rituximab in advanced-stage disease has suggested the use of R-CHOP with or without radiation therapy but its use is only supported by retrospective comparisons.[4][Level of evidence C2]
- R-CHOP (four to six cycles).
- R-CHOP (three to six cycles) + IF-XRT.
Evidence (R-CHOP with or without IF-XRT):
- In a randomized prospective trial of 334 patients with nonbulky (≤7 cm) stage I or stage II DLBCL, after receiving four to six cycles of R-CHOP-14 (R-CHOP delivered every 2 weeks), patients were randomly assigned to receive or not receive 40 Gy of radiation therapy.[5]
- After a median follow-up of 64 months, the 5-year event-free survival (89%–92%, P = .18) and 5-year overall survival (OS) (92%–96%, P = .32) were the same.[5][Level of evidence A1]
Similar to the results of randomized studies of radiation therapy in the prerituximab era, radiation therapy can be deferred in nonbulky early-stage patients. For patients unable to tolerate prolonged-course chemotherapy, three cycles of R-CHOP plus radiation therapy has produced equivalent results based on single-arm retrospective trials.[4]
- In a randomized prospective trial (NCT00278421) of 592 patients younger than 60 years with nonbulky (<7.5 cm) stage I or stage II DLBCL, patients were randomly assigned to receive either four or six cycles of R-CHOP (with an extra two cycles of rituximab after four cycles).[6]
- With a median follow-up of 66 months, the 3-year progression-free survival (PFS) rate was 96% (95% CI, 94%−99%)for patients who received four cycles of R-CHOP, which was 3% better (lower limit of one-sided 95% CI was zero) than the PFS rate for patients who received six cycles, establishing noninferiority for the four-cycle regimen.[6][Level of evidence B1]
- A retrospective analysis at Memorial Sloan Kettering Cancer Center between 2001 and 2015 included 341 patients with stage I disease. The analysis found that 66% of patients had an extranodal presentation. After R-CHOP (or a similar regimen), with or without radiation therapy, the 5-year disease-free survival rate was 77%, and the 5-year OS rate was 94%.[7][Level of evidence C3] A multivariate analysis suggested that radiation therapy may improve outcomes for patients with extranodal disease that is positron emission tomography (PET)–positive at the end of therapy. This hypothesis needs confirmation in a prospective randomized trial.[7]
In summary, for patients with favorable prognosis nonbulky (<7 cm) stage I or stage II DLBCL, four cycles of R-CHOP is sufficient. For patients with unfavorable prognosis, six cycles of R-CHOP or three cycles of R-CHOP and 40 Gy of radiation therapy can be used. Early-stage patients with bulky disease (>7.5 cm) have not been studied in randomized trials; combined-modality therapy with R-CHOP for four to six cycles plus radiation therapy is usually chosen. Although a retrospective study suggested that patients with stage I extranodal disease and a positive PET scan at the end of therapy may benefit from radiation therapy, this hypothesis must be confirmed in a prospective randomized trial.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
- Reyes F, Lepage E, Ganem G, et al.: ACVBP versus CHOP plus radiotherapy for localized aggressive lymphoma. N Engl J Med 352 (12): 1197-205, 2005.
- Ketterer N, Coiffier B, Thieblemont C, et al.: Phase III study of ACVBP versus ACVBP plus rituximab for patients with localized low-risk diffuse large B-cell lymphoma (LNH03-1B). Ann Oncol 24 (4): 1032-7, 2013.
- Persky DO, Unger JM, Spier CM, et al.: Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 26 (14): 2258-63, 2008.
- Lamy T, Damaj G, Soubeyran P, et al.: R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 131 (2): 174-181, 2018.
- Poeschel V, Held G, Ziepert M, et al.: Four versus six cycles of CHOP chemotherapy in combination with six applications of rituximab in patients with aggressive B-cell lymphoma with favourable prognosis (FLYER): a randomised, phase 3, non-inferiority trial. Lancet 394 (10216): 2271-2281, 2019.
- Bobillo S, Joffe E, Lavery JA, et al.: Clinical characteristics and outcomes of extranodal stage I diffuse large B-cell lymphoma in the rituximab era. Blood 137 (1): 39-48, 2021.
Treatment of Aggressive, Noncontiguous Stage II / III / IV NHL
The treatment of choice for patients with advanced stages of aggressive non-Hodgkin lymphoma (NHL) is combination chemotherapy, either alone or supplemented by local-field radiation therapy.[1]
The following drug combinations are referred to in this section:
- R-CHOP: rituximab + cyclophosphamide + doxorubicin + vincristine + prednisone.
- R-ACVBP: rituximab, an anti–CD20 monoclonal antibody, + doxorubicin + cyclophosphamide + vindesine + bleomycin + prednisone.
Treatment Options for Aggressive, Noncontiguous Stage II/III/IV NHL
Treatment options for aggressive, noncontiguous stage II/III/IV NHL include the following:
- R-CHOP.
- Other combination chemotherapy.
- Bone marrow transplantation (BMT) or stem cell transplantation (SCT) (under clinical evaluation).
- Radiation therapy consolidation to sites of bulky disease (under clinical evaluation).
R-CHOP
The following studies established R-CHOP as the standard regimen for newly diagnosed patients with diffuse large B-cell lymphoma (DLBCL).[2] Dose intensification of R-CHOP by a 14-day versus a 21-day cycle did not result in improved outcomes.[3]
Evidence (R-CHOP):
- R-CHOP showed improvement in event-free survival (EFS) and overall survival (OS) compared with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) alone in 399 advanced-stage patients with DLBCL older than 60 years (EFS rate, 57% vs. 38%; P = .002, and OS rate, 70% vs. 57%; P = .007 at 2 years).[4][Level of evidence A1] At a median follow-up of 10 years, the OS rate of patients who received R-CHOP compared with patients who received CHOP was 44% versus 28% (P < .0001).[5]
- Similarly, for 326 evaluable patients younger than 61 years, R-CHOP showed improvement in EFS and OS compared with CHOP alone (EFS rate, 79% vs. 59%, P = .001, and OS rate, 93% vs. 84%, P = .001 at 3 years).[6][Level of evidence A1]
- A randomized study (DSHNHL-1999-1A [NCT00052936]) of 1,222 patients older than 60 years compared R-CHOP given every 2 weeks for six or eight cycles with CHOP given every 2 weeks for six or eight cycles.[7] With a median follow-up of 72 months, the EFS favored R-CHOP given every 2 weeks for six or eight cycles (EFS rate at 6 years, 74% vs. 56%; P < .0001). The OS favored R-CHOP for only six cycles because of increased toxicity in the eight-cycle arm (OS rate at 6 years, 90% vs. 80%; P = .0004).[7][Level of evidence A1] There was no comparison with standard R-CHOP or CHOP given every 3 weeks.
- A trial (NCT00140595) of 380 patients younger than 60 years with DLBCL and an age-adjusted International Prognostic Index (IPI) rating of 1 randomly assigned treatment of patients to ACVBP and R-ACVBP plus consolidation with methotrexate, ifosfamide, etoposide, and cytarabine versus CHOP and rituximab.[8] With a median follow-up of 44 months, 3-year OS rates favored R-ACVBP (92% vs. 84%; hazard ratio [HR], 0.44; 95% confidence interval [CI], 0.28–0.81, P = .007).[8][Level of evidence A1] The significantly worse toxicities with R-ACVBP, the narrow target population (<60 years with either elevated lactate dehydrogenase (LDH) or stage III-stage IV disease, but not both), and the lack of a confirmatory trial may inhibit adoption of R-ACVBP as a new standard of care.[9]
Modifications to R-CHOP to achieve improved efficacy continue to be explored in clinical trials. For over 2 decades, no modification has replaced R-CHOP as the worldwide standard induction therapy.[10]
Pola-R-CHP
R-CHOP has been compared to Pola-R-CHP (polatuzumab vedotin, rituximab, cyclophosphamide, doxorubicin, and prednisone). Polatuzumab is an antibody-drug conjugate composed of an anti-CD79B monoclonal antibody attached to vedotin (monomethyl auristatin E), a microtubule inhibitor.
Evidence (Pola-R-CHP):
- A provocative, prospective, randomized study of 879 patients with previously untreated DLBCL compared R-CHOP with Pola-R-CHP.[11] Polatuzumab vedotin was substituted for vincristine to mitigate neurological toxicity.
- At a median follow-up of 28.2 months, the 2-year PFS rate was significantly higher in the Pola-R-CHP group than in the R-CHOP group: 76.7% (95% CI, 72.7%–80.0%) versus 70.2% (95% CI, 65.8%–74.6%) (HR, 0.73; 95% CI, 0.57–0.95; P = .02).[11][Level of evidence B1]
- The 2-year OS rate was 88.7% (95% CI, 85.7%–91.6%) for patients who received Pola-R-CHP and 88.6% (95% CI, 85.6%–91.6%) for patients who received R-CHOP (HR, 0.94; 95% CI, 0.65–1.37; P = .75).
The follow-up interval was too short to establish whether the 6% improvement in the PFS rate will plateau or improve after 2 years, and there is no evidence of OS advantage. Nonetheless, physician advisory panels are considering whether this represents a new standard regimen to replace R-CHOP. The new regimen is more than twice the cost of R-CHOP using acquisition prices in 2022.
There is no validated trial for interim positron emission tomography–based treatment intensification.[12] R-CHOP has curative potential even in patients older than 80 years who are frail and require reduced dosage of R-CHOP components. In a retrospective review of 239 patients, the 5-year cause-specific survival rate was 48% (95% CI, 41%−55%).[13][Level of evidence C3]
Less than 10% of patients with DLBCL present with a concurrent indolent lymphoma at diagnosis, and these are predominantly of germinal center B-cell phenotype. A retrospective review of 1,324 patients showed similar EFS (HR, 1.19) and OS (HR, 1.09).[14][Level of evidence C3] For 847 patients who were treated with R-CHOP and free of disease 24 months after therapy, the rate of indolent lymphoma relapse by 5 years was higher with a concurrent diagnosis of follicular lymphoma (7.4% vs. 2.1% at 5 years, P < .01) and with a germinal center B-cell phenotype (3.9% vs. 0.0% at 5 years, P = .02).[15]
Stage IE or IIE gastric DLBCL
Four case series involving more than 100 patients with stage IE or IIE disease (with or without associated mucosa-associated lymphatic tissue) and with positive Helicobacter pylori infection reported that more than 50% of patients attained a durable complete remission after appropriate antibiotic therapy to eradicate H. pylori.[16,17,18,19][Level of evidence C3]
Prognostic factors
The National Comprehensive Cancer Network IPI for aggressive NHL (diffuse large cell lymphoma) identifies the following five significant risk factors prognostic of OS and their associated risk scores:[20]
- Age.
- <40 years: 0.
- 41–60 years: 1.
- 61–75 years: 2.
- >75 years: 3.
- Stage III/IV: 1.
- Performance status 2/3/4: 1.
- Serum LDH.
- Normalized: 0.
- >1x to 3x: 1.
- >3x: 2.
- Number of extranodal sites ≥2: 1.
Risk scores:
- Low (0 or 1): 5-year OS, 96%; progression-free survival (PFS), 91%.
- Low intermediate (2 or 3): 5-year OS, 82%; PFS, 74%.
- High intermediate (4 or 5): 5-year OS, 64%; PFS, 51%.
- High (>6): 5-year OS, 33%; PFS, 30%.
Age-adjusted and stage-adjusted modifications of this IPI are used for younger patients with localized disease.[21] Shorter intervals of time between diagnosis and treatment appear to be a surrogate for poor prognostic biological factors.[22]
The BCL2 gene and rearrangement of the MYC gene or dual overexpression of the MYC gene, or both, confer a particularly poor prognosis.[23,24,25,26] Patients at high risk of relapse may be considered for clinical trials.[27] Molecular profiles of gene expression using DNA microarrays may help to stratify patients in the future for therapies directed at specific targets and to better predict survival after standard chemotherapy.[28]
Treatment of tumor lysis syndrome
Patients with bulky and extensive lymphadenopathy and elevations of serum uric acid and LDH are at increased risk of tumor lysis syndrome resulting in metabolic derangements such as hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia, and subsequent acute renal failure.[29] Treatment options include: alkaline hydration, allopurinol, and rasburicase, a recombinant urate oxidase.[30]
CNS prophylaxis
The CNS-IPI tool predicts which patients have a CNS relapse risk exceeding 10%. It was developed by the German Lymphoma Study Group and validated by the British Columbia Cancer Agency database. The presence of four to six of the CNS-IPI risk factors (age >60 years, performance status ≥2, elevated LDH, stage III or IV disease, >1 extranodal site, or involvement of the kidneys or adrenal glands) was used to define a high-risk group for CNS recurrence (a 12% risk of CNS involvement by 2 years).[31]
CNS prophylaxis (usually with four to six doses of intrathecal methotrexate) is often recommended for patients with testicular involvement.[32,33,34][Level of evidence C3] A retrospective analysis of the German RICOVER studies compared intrathecal methotrexate with no prophylaxis in patients with DLBCL. This study was completed during the R-CHOP treatment era. With the possible exception of patients with testicular involvement, the analysis showed that intrathecal methotrexate did not reduce the risk of CNS disease.[35][Level of evidence C3] Some clinicians employ high-dose intravenous (IV) methotrexate (usually four doses) as an alternative to intrathecal therapy because drug delivery is improved and patient morbidity is decreased.[36] Five retrospective studies and one network meta-analysis evaluating high-dose methotrexate alone in patients with high-risk DLBCL also showed no improvement in CNS relapse rate.[37,38,39,40,41,42][Level of evidence C3] Patients deemed at high risk for CNS relapse (e.g., patients with four to six CNS-IPI risk factors) may receive intrathecal methotrexate or high-dose IV methotrexate, but the lack of confirmatory randomized studies calls this standard into question and shows an urgent need for better therapeutics verified in clinical trials. Although patients with testicular involvement or kidney/adrenal involvement are considered an exception, there is only anecdotal benefit from intrathecal or high-dose IV methotrexate in reducing CNS recurrence.[32,33,34][Level of evidence C3]
The addition of rituximab to CHOP-based regimens has significantly reduced the risk of CNS relapse in retrospective analyses.[35,43][Level of evidence C3] Patients with CNS dissemination at diagnosis or at relapse usually receive rituximab and high doses of methotrexate and/or cytarabine followed by autologous stem cell transplantation (SCT), but this approach has not been assessed in randomized trials.[44,45][Level of evidence C3]
BMT or SCT
Several randomized prospective trials evaluated the role of autologous BMT or SCT consolidation versus chemotherapy alone in patients with diffuse large cell lymphoma in first remission.[46,47,48,49,50,51,52,53]; [54,55,56][Level of evidence A1] Although some of these trials demonstrated significant increases in EFS (by 10% to 20%) among patients who received high-dose therapy, significant differences in OS could not be demonstrated prospectively in any of the series.
Retrospective analyses of high-intermediate (two risk factors) or high-risk (more than three risk factors) patients as defined by IPI suggest improved survival with BMT in two of the trials.[47,53] These studies do not establish that high-dose consolidation is of value to patients with aggressive lymphoma who are truly at high risk of relapse, and they also demonstrate that EFS may be a poor surrogate for OS for these patients.[57]
Radiation therapy consolidation to sites of bulky disease
After R-CHOP induction chemotherapy (or similar regimens), the addition of involved-field radiation therapy to sites of initial bulky disease (≥5–10 cm) or to extralymphatic sites remains controversial.[58,59,60] Increased risks, such as long-term toxicities (e.g., second malignancies), must be considered.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Shankland KR, Armitage JO, Hancock BW: Non-Hodgkin lymphoma. Lancet 380 (9844): 848-57, 2012.
- Coiffier B: State-of-the-art therapeutics: diffuse large B-cell lymphoma. J Clin Oncol 23 (26): 6387-93, 2005.
- Cunningham D, Hawkes EA, Jack A, et al.: Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet 381 (9880): 1817-26, 2013.
- Coiffier B, Lepage E, Briere J, et al.: CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346 (4): 235-42, 2002.
- Coiffier B, Thieblemont C, Van Den Neste E, et al.: Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 116 (12): 2040-5, 2010.
- Pfreundschuh M, Trümper L, Osterborg A, et al.: CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 7 (5): 379-91, 2006.
- Pfreundschuh M, Kuhnt E, Trümper L, et al.: CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 12 (11): 1013-22, 2011.
- Récher C, Coiffier B, Haioun C, et al.: Intensified chemotherapy with ACVBP plus rituximab versus standard CHOP plus rituximab for the treatment of diffuse large B-cell lymphoma (LNH03-2B): an open-label randomised phase 3 trial. Lancet 378 (9806): 1858-67, 2011.
- Casasnovas RO, Ysebaert L, Thieblemont C, et al.: FDG-PET-driven consolidation strategy in diffuse large B-cell lymphoma: final results of a randomized phase 2 study. Blood 130 (11): 1315-1326, 2017.
- Nowakowski GS, Hong F, Scott DW, et al.: Addition of Lenalidomide to R-CHOP Improves Outcomes in Newly Diagnosed Diffuse Large B-Cell Lymphoma in a Randomized Phase II US Intergroup Study ECOG-ACRIN E1412. J Clin Oncol 39 (12): 1329-1338, 2021.
- Tilly H, Morschhauser F, Sehn LH, et al.: Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med 386 (4): 351-363, 2022.
- Dührsen U, Müller S, Hertenstein B, et al.: Positron Emission Tomography-Guided Therapy of Aggressive Non-Hodgkin Lymphomas (PETAL): A Multicenter, Randomized Phase III Trial. J Clin Oncol 36 (20): 2024-2034, 2018.
- Gobba S, Moccia AA, Gulden-Sala W, et al.: Outcome of patients older than 80 years with diffuse large B-cell lymphoma (DLBCL) treated with "standard" immunochemotherapy: A large retrospective study from 4 institutions. Hematol Oncol 36 (1): 84-92, 2018.
- Wang Y, Link BK, Witzig TE, et al.: Impact of concurrent indolent lymphoma on the clinical outcome of newly diagnosed diffuse large B-cell lymphoma. Blood 134 (16): 1289-1297, 2019.
- Wang Y, Farooq U, Link BK, et al.: Late Relapses in Patients With Diffuse Large B-Cell Lymphoma Treated With Immunochemotherapy. J Clin Oncol 37 (21): 1819-1827, 2019.
- Morgner A, Miehlke S, Fischbach W, et al.: Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 19 (7): 2041-8, 2001.
- Chen LT, Lin JT, Shyu RY, et al.: Prospective study of Helicobacter pylori eradication therapy in stage I(E) high-grade mucosa-associated lymphoid tissue lymphoma of the stomach. J Clin Oncol 19 (22): 4245-51, 2001.
- Chen LT, Lin JT, Tai JJ, et al.: Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst 97 (18): 1345-53, 2005.
- Kuo SH, Yeh KH, Wu MS, et al.: Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood 119 (21): 4838-44; quiz 5057, 2012.
- Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
- Møller MB, Christensen BE, Pedersen NT: Prognosis of localized diffuse large B-cell lymphoma in younger patients. Cancer 98 (3): 516-21, 2003.
- Maurer MJ, Ghesquières H, Link BK, et al.: Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36 (16): 1603-1610, 2018.
- Cuccuini W, Briere J, Mounier N, et al.: MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood 119 (20): 4619-24, 2012.
- Johnson NA, Slack GW, Savage KJ, et al.: Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30 (28): 3452-9, 2012.
- Green TM, Young KH, Visco C, et al.: Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30 (28): 3460-7, 2012.
- Horn H, Ziepert M, Becher C, et al.: MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121 (12): 2253-63, 2013.
- Canellos GP: CHOP may have been part of the beginning but certainly not the end: issues in risk-related therapy of large-cell lymphoma. J Clin Oncol 15 (5): 1713-6, 1997.
- Sha C, Barrans S, Cucco F, et al.: Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37 (3): 202-212, 2019.
- Coiffier B, Altman A, Pui CH, et al.: Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol 26 (16): 2767-78, 2008.
- Cortes J, Moore JO, Maziarz RT, et al.: Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone--results of a multicenter phase III study. J Clin Oncol 28 (27): 4207-13, 2010.
- Schmitz N, Zeynalova S, Nickelsen M, et al.: CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 34 (26): 3150-6, 2016.
- Zucca E, Conconi A, Mughal TI, et al.: Patterns of outcome and prognostic factors in primary large-cell lymphoma of the testis in a survey by the International Extranodal Lymphoma Study Group. J Clin Oncol 21 (1): 20-7, 2003.
- Vitolo U, Chiappella A, Ferreri AJ, et al.: First-line treatment for primary testicular diffuse large B-cell lymphoma with rituximab-CHOP, CNS prophylaxis, and contralateral testis irradiation: final results of an international phase II trial. J Clin Oncol 29 (20): 2766-72, 2011.
- Cheah CY, Wirth A, Seymour JF: Primary testicular lymphoma. Blood 123 (4): 486-93, 2014.
- Boehme V, Schmitz N, Zeynalova S, et al.: CNS events in elderly patients with aggressive lymphoma treated with modern chemotherapy (CHOP-14) with or without rituximab: an analysis of patients treated in the RICOVER-60 trial of the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL). Blood 113 (17): 3896-902, 2009.
- Glantz MJ, Cole BF, Recht L, et al.: High-dose intravenous methotrexate for patients with nonleukemic leptomeningeal cancer: is intrathecal chemotherapy necessary? J Clin Oncol 16 (4): 1561-7, 1998.
- Puckrin R, El Darsa H, Ghosh S, et al.: Ineffectiveness of high-dose methotrexate for prevention of CNS relapse in diffuse large B-cell lymphoma. Am J Hematol 96 (7): 764-771, 2021.
- Jeong H, Cho H, Kim H, et al.: Efficacy and safety of prophylactic high-dose MTX in high-risk DLBCL: a treatment intent-based analysis. Blood Adv 5 (8): 2142-2152, 2021.
- Orellana-Noia VM, Reed DR, McCook AA, et al.: Single-route CNS prophylaxis for aggressive non-Hodgkin lymphomas: real-world outcomes from 21 US academic institutions. Blood 139 (3): 413-423, 2022.
- Lewis KL, Jakobsen LH, Villa D, et al.: High-dose methotrexate is not associated with reduction in CNS relapse in patients with aggressive B-cell lymphoma: an international retrospective study of 2300 high-risk patients. [Abstract] Blood 138 (Suppl 1): A-181, 2021.
- Wilson MR, Eyre TA, Kirkwood AA, et al.: Timing of high-dose methotrexate CNS prophylaxis in DLBCL: a multicenter international analysis of 1384 patients. Blood 139 (16): 2499-2511, 2022.
- Haddad PA, Hammoud D, Gallagher KM: Effective central nervous system (CNS) prophylaxis chemotherapy approach in high risk diffuse large B-cell lymphoma (DLBCL): a network meta-analysis. [Abstract] Blood 138 (Suppl 1): A-1461, 2021.
- Villa D, Connors JM, Shenkier TN, et al.: Incidence and risk factors for central nervous system relapse in patients with diffuse large B-cell lymphoma: the impact of the addition of rituximab to CHOP chemotherapy. Ann Oncol 21 (5): 1046-52, 2010.
- Ferreri AJ, Donadoni G, Cabras MG, et al.: High Doses of Antimetabolites Followed by High-Dose Sequential Chemoimmunotherapy and Autologous Stem-Cell Transplantation in Patients With Systemic B-Cell Lymphoma and Secondary CNS Involvement: Final Results of a Multicenter Phase II Trial. J Clin Oncol 33 (33): 3903-10, 2015.
- Schmitz N, Wu HS: Advances in the Treatment of Secondary CNS Lymphoma. J Clin Oncol 33 (33): 3851-3, 2015.
- Haioun C, Lepage E, Gisselbrecht C, et al.: Survival benefit of high-dose therapy in poor-risk aggressive non-Hodgkin's lymphoma: final analysis of the prospective LNH87-2 protocol--a groupe d'Etude des lymphomes de l'Adulte study. J Clin Oncol 18 (16): 3025-30, 2000.
- Haioun C, Lepage E, Gisselbrecht C, et al.: Benefit of autologous bone marrow transplantation over sequential chemotherapy in poor-risk aggressive non-Hodgkin's lymphoma: updated results of the prospective study LNH87-2. Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 15 (3): 1131-7, 1997.
- Santini G, Salvagno L, Leoni P, et al.: VACOP-B versus VACOP-B plus autologous bone marrow transplantation for advanced diffuse non-Hodgkin's lymphoma: results of a prospective randomized trial by the non-Hodgkin's Lymphoma Cooperative Study Group. J Clin Oncol 16 (8): 2796-802, 1998.
- Gianni AM, Bregni M, Siena S, et al.: High-dose chemotherapy and autologous bone marrow transplantation compared with MACOP-B in aggressive B-cell lymphoma. N Engl J Med 336 (18): 1290-7, 1997.
- Kluin-Nelemans HC, Zagonel V, Anastasopoulou A, et al.: Standard chemotherapy with or without high-dose chemotherapy for aggressive non-Hodgkin's lymphoma: randomized phase III EORTC study. J Natl Cancer Inst 93 (1): 22-30, 2001.
- Gisselbrecht C, Lepage E, Molina T, et al.: Shortened first-line high-dose chemotherapy for patients with poor-prognosis aggressive lymphoma. J Clin Oncol 20 (10): 2472-9, 2002.
- Martelli M, Gherlinzoni F, De Renzo A, et al.: Early autologous stem-cell transplantation versus conventional chemotherapy as front-line therapy in high-risk, aggressive non-Hodgkin's lymphoma: an Italian multicenter randomized trial. J Clin Oncol 21 (7): 1255-62, 2003.
- Milpied N, Deconinck E, Gaillard F, et al.: Initial treatment of aggressive lymphoma with high-dose chemotherapy and autologous stem-cell support. N Engl J Med 350 (13): 1287-95, 2004.
- Betticher DC, Martinelli G, Radford JA, et al.: Sequential high dose chemotherapy as initial treatment for aggressive sub-types of non-Hodgkin lymphoma: results of the international randomized phase III trial (MISTRAL). Ann Oncol 17 (10): 1546-52, 2006.
- Stiff PJ, Unger JM, Cook JR, et al.: Autologous transplantation as consolidation for aggressive non-Hodgkin's lymphoma. N Engl J Med 369 (18): 1681-90, 2013.
- Chiappella A, Martelli M, Angelucci E, et al.: Rituximab-dose-dense chemotherapy with or without high-dose chemotherapy plus autologous stem-cell transplantation in high-risk diffuse large B-cell lymphoma (DLCL04): final results of a multicentre, open-label, randomised, controlled, phase 3 study. Lancet Oncol 18 (8): 1076-1088, 2017.
- Shipp MA, Abeloff MD, Antman KH, et al.: International Consensus Conference on high-dose therapy with hematopoietic stem-cell transplantation in aggressive non-Hodgkin's lymphomas: report of the jury. Ann Oncol 10 (1): 13-9, 1999.
- Held G, Murawski N, Ziepert M, et al.: Role of radiotherapy to bulky disease in elderly patients with aggressive B-cell lymphoma. J Clin Oncol 32 (11): 1112-8, 2014.
- Kahl BS: Bulky aggressive B-cell lymphoma: to radiate or not to radiate--that is the question. J Clin Oncol 32 (11): 1097-8, 2014.
- Phan J, Mazloom A, Medeiros LJ, et al.: Benefit of consolidative radiation therapy in patients with diffuse large B-cell lymphoma treated with R-CHOP chemotherapy. J Clin Oncol 28 (27): 4170-6, 2010.
Treatment of Aggressive, Recurrent NHL
Treatment Options for Aggressive, Recurrent NHL
In a retrospective review of multiple international trials, 636 patients were identified as having refractory diffuse large B-cell lymphoma (DLBCL), which was defined as progression or stable disease during or just at completion of full-course chemotherapy or relapse within 1 year after autologous stem cell transplantation (SCT).[1] With subsequent therapy the objective response rate was 26%, complete response (CR) rate was 7%, median overall survival (OS) was 6.3 months, and only 20% of patients were alive at 2 years. Even with reinduction chemotherapy with planned autologous SCT, outcomes remain poor.[2]
Treatment options for aggressive, recurrent non-Hodgkin lymphoma (NHL) include the following:
- Chimeric antigen receptor (CAR) T-cell therapy for primary refractory disease or relapse within one year.
- Bone marrow transplantation (BMT)/SCT consolidation.
- CAR T-cell therapy for relapse after autologous SCT.
- Tafasitamab plus lenalidomide.
- Rituximab plus lenalidomide.
- Polatuzumab vedotin plus rituximab and bendamustine.
- Loncastuximab tesirine.
- Bispecific T-cell engagers.
- Palliative radiation therapy.
CAR T-cell therapy for primary refractory disease or relapse within one year
Patients with DLBCL who relapse during or within 2 months of receiving R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy have primary refractory disease. Any patient with disease relapse within 1 year of R-CHOP chemotherapy or with primary refractory disease has a poor prognosis even with reinduction using chemoimmunotherapy followed by autologous SCT.[1,2] Patients who received CAR T-cell therapy had a 40% to 50% 3-year progression-free survival (PFS) rate with a 40-month follow-up, a result equivalent retrospectively to autologous SCT in bone marrow registries.[3,4,5,6]
Three randomized trials compared chemoimmunotherapy followed by autologous SCT with CAR T-cell therapy with or without bridging chemoimmunotherapy for patients with high-risk relapsed disease defined as primary refractory disease or relapse within 12 months of initial R-CHOP therapy.
Evidence (CAR T-cell therapy):
- A prospective randomized trial included 359 patients with primary refractory disease or relapse within 12 months of initial R-CHOP chemotherapy. Patients received the CAR T-cell therapy axicabtagene ciloleucel with only bridging steroids or second-line chemoimmunotherapy (usually R-ICE [rituximab, ifosfamide, etoposide, and carboplatin] or R-DHAP [rituximab, dexamethasone, high-dose cytarabine, and cisplatin]) followed by autologous SCT.[7]
- With a median follow-up of 24.9 months, the 2-year event-free survival (EFS) rate was 40.5% for patients who received axicabtagene ciloleucel and 16.3% for patients who received chemoimmunotherapy followed by autologous SCT (hazard ratio [HR], 0.398; 95% confidence interval [CI], 0.308–0.514; P < .0001).[7][Level of evidence B1]
- The OS was not significantly better at 2 years, with an HR of 0.73 (95% CI, 0.53–1.01; P = .027).
- On the chemoimmunotherapy arm of the study, 64% of patients never received autologous SCT because of inadequate response, progression, or death.
- Clinically meaningful and statistically significant differences in quality of life were obtained in the CAR T-cell arm at day 100 and day 150, compared with the standard of care.[8][Level of evidence A2]
- Grade 3 or 4 cytokine release syndrome occurred in 6% of patients and grade 3 or 4 neurotoxicity occurred in 21% of patients.
- A prospective randomized trial included 184 patients with primary refractory disease or relapse within 12 months of initial R-CHOP chemotherapy. Patients received the CAR T-cell therapy lisocabtagene maraleucel, with 63% of patients receiving bridging therapy or second-line chemoimmunotherapy followed by autologous SCT.[9]
- With a median follow-up of 6.2 months, the median PFS was 14.8 months for patients who received lisocabtagene maraleucel and 5.7 months for patients who received chemoimmunotherapy followed by autologous SCT (HR, 0.41; P = .001).[9][Level of evidence B1]
- On the chemoimmunotherapy arm of the study, 53% of patients never received autologous SCT because of inadequate response, progression, or death.[9]
- Grade 3 cytokine release syndrome occurred in 1% of patients, and grade 3 neurotoxicity occurred in 4% of patients. There were no grade 4 or 5 occurrences.
- A prospective randomized trial included 322 patients with primary refractory disease or relapse within 12 months of initial R-CHOP chemotherapy. Patients received the CAR T-cell therapy tisagenlecleucel, with most patients receiving bridging therapy to achieve response, or second-line chemoimmunotherapy followed by autologous SCT.[10]
- There was no difference in EFS for patients in either arm (HR, 1.07; 95% CI, 0.82–1.40; P = .69).[10][Level of evidence B1]
- In the CAR T-cell therapy arm, 48% of patients received two or more cycles of chemoimmunotherapy as part of bridging therapy. This approach to bridging therapy may have led to an unacceptable number of cases of progressive disease.
In summary:
- For patients with high-risk relapsing DLBCL with primary refractory disease or relapse within 12 months of R-CHOP-based chemotherapy, axicabtagene ciloleucel and lisocabtagene maraleucel are superior induction regimens compared with chemoimmunotherapy with regimens like R-ICE, R-DHAP, and R-GDP (rituximab, gemcitabine, dexamethasone, and cisplatin).
- The interval until patients receive CAR T cells must be minimized, optimally by using only steroids, eliminating bridging chemoimmunotherapy, and infusing the CAR T-cell product as quickly as possible.
- The preference for CAR T-cell therapy over chemoimmunotherapy followed by autologous SCT does not apply to patients who relapse more than 12 months after R-CHOP therapy.
- The American Society of Clinical Oncology (ASCO) has compiled guidelines for the management of adverse events in patients treated with CAR T-cell therapy.[11]
Bone marrow/stem cell transplantation consolidation
BMT consolidation is a treatment for patients whose lymphoma has relapsed.[12] Preliminary studies indicate that approximately 20% to 40% of patients will have a long-term disease-free status, but the precise percentage depends on patient selection and the specific treatment used. Preparative drug regimens have varied; some investigators also use total-body irradiation. Similar success has been achieved using autologous marrow, with or without marrow purging, and allogeneic marrow.[13,14,15,16,17]
Evidence (BMT):
- In a prospective randomized study, (EORTC-PARMA), 215 patients in first or second relapse of aggressive lymphoma, younger than 60 years, and with no bone marrow or central nervous system involvement, were given two cycles of intensive combination chemotherapy. The 109 patients who responded were randomly assigned to receive four more cycles of chemotherapy and involved-field radiation therapy (IF-XRT) versus autologous BMT followed by IF-XRT. With a 5-year median follow-up, the EFS rate was significantly improved with transplantation (46% vs. 12%). The OS rate was also significantly better with transplantation (53% vs. 32%).[18][Level of evidence A1] Salvage BMT was unsuccessful for patients on the nontransplant arm whose disease relapsed.
In general, patients who responded to initial therapy and who responded to conventional therapy for relapse before the BMT have had the best results.[19]
- In a prospective trial, patients who relapsed late (>12 months after diagnosis) had better OS than patients who relapsed earlier (the 8-year survival rate was 29% vs. 13%, P = .001).[20][Level of evidence C1]
Peripheral SCT has yielded results equivalent to standard autologous SCT.[21,22] Even patients who never experienced complete remission with conventional chemotherapy may have prolonged PFS (31% at 5 years) after high-dose chemotherapy and hematopoietic SCT if they retain chemosensitivity to reinduction therapy.[23][Level of evidence C2] Some patients who relapse after a previous autologous SCT can have durable remissions after myeloablative or nonmyeloablative allogeneic SCT.[24,25]; [26][Level of evidence C3] Reduced-intensity conditioning for allogeneic SCT typically involves fludarabine plus busulfan or fludarabine plus cyclophosphamide, with or without 2 Gy of total-body irradiation.[27]
Evidence (peripheral SCT):
- In a randomized prospective trial, 396 patients with DLBCL in first relapse or who were refractory to first-line therapy received either R-ICE or R-DHAP followed by autologous SCT.[28]
- There was no difference in 3-year EFS or OS.[28][Level of evidence A1]
- In a randomized prospective trial, 619 patients with relapsed or refractory aggressive lymphoma received either R-DHAP or R-GDP followed by autologous SCT.[29]
- At a median follow-up of 53 months, there was no difference in EFS or OS, but patients who received R-GDP reported less toxicity.[29][Level of evidence A3]
CAR T-cell therapy for relapse after autologous SCT
In the event of disease relapse after autologous SCT, many patients receive consolidation with CAR T-cell therapy.
Multiple trials describe patients with refractory large B-cell lymphoma who underwent an infusion of T cells that were engineered to express a CAR to target the CD19 antigen expressed on the malignant B cells using three different constructs: axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel.[30,31,32,33,34] Each study reported a CR rate of 50% to 60% and a 2-year OS rate of 40% to 50%, but the long-term durability of response has yet to be determined in these highly-selected patients.[30,31,32][Level of evidence C3] This therapy is an option for patients with otherwise refractory or resistant disease. These results have been verified off-study in two reports that included 397 patients treated after U.S. Food and Drug Administration (FDA) approval.[35,36][Level of evidence C3] The highest risk patients who respond adequately may receive a subsequent allogeneic SCT consolidation in some cases if eligible.
ASCO has compiled guidelines for the management of adverse events in patients treated with CAR T-cell therapy.[11]
Tafasitamab plus lenalidomide
Evidence (tafasitamab plus lenalidomide):
- In a phase II study, 80 patients with relapsed or refractory DLBCL were treated with tafasitamab, a CD19-directed humanized monoclonal antibody combined with lenalidomide.[37]
- The CR rate was 43% and the objective response rate was 60%.
The FDA approved the combination of tafasitamab and lenalidomide for patients with relapsed or refractory DLBCL.[37][Level of evidence C3] It is unclear if using this CD19-directed therapy would interfere with consolidation using CD19-CAR T cells.
Rituximab plus lenalidomide
Evidence (rituximab plus lenalidomide):
- In two phase II trials, 49 patients showed a 19% to 35% overall response rate to lenalidomide with rituximab.[38,39][Level of evidence C3]
Polatuzumab vedotin plus rituximab and bendamustine
Polatuzumab vedotin is a CD79b-directed monoclonal antibody conjugated to the cytotoxic agent vedotin (an antibody-drug conjugate).
Evidence (polatuzumab vedotin plus rituximab and bendamustine):
- In a randomized, prospective trial, 80 patients with relapsed or refractory DLBCL were treated with either polatuzumab vedotin combined with bendamustine and rituximab (BR) or BR alone, with a primary end point of complete response.[40]
- The CR rate by positron emission tomography−computed tomography scan was 40% for the polatuzumab vedotin-BR combination, compared with 18% for BR alone (P = .026).[40]
- Similarly, the median PFS was higher for patients who received the polatuzumab vedotin combination (9.5 months) than for the patients who received BR alone (3.7 months) (HR, 0.36; 95% CI, 0.21−0.63; P < .001); the OS was 12.4 months for patients who received the polatuzumab vedotin combination versus 4.7 months for the patients who received BR alone (HR, 0.42; 95% CI, 0.24−0.75; P = .002).[40][Level of evidence C1]
The FDA approved the combination of polatuzumab vedotin and BR for patients with relapsed or refractory DLBCL.
Loncastuximab tesirine
Loncastuximab tesirine is a CD19-directed antibody conjugated to a pyrrolobenzodiazepine dimer cytotoxin (an antibody-drug conjugate).[41]
Evidence (loncastuximab tesirine):
- A phase I and subsequent phase II trial included 184 patients with relapsed or refractory disease.
- The overall response rate was 48.3% (95% CI, 39.9%–56.7%) and the CR rate was 24%.[42,43][Level of evidence C3]
Bispecific T-cell engagers
Bispecific T-cell engagers bind to CD20 (or CD19) and to CD3 to direct T cells to eliminate malignant B cells.[44] As with CAR T-cell therapy, almost one-half of patients who receive this therapy experience cytokine release syndrome.
Palliative radiation therapy
In general, patients with aggressive lymphoma who relapse with indolent histology will benefit from palliative therapy.[45] Palliation may be achieved with very low-dose (4 Gy) IF-XRT for patients with indolent and aggressive relapsed disease.[46]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Crump M, Neelapu SS, Farooq U, et al.: Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 130 (16): 1800-1808, 2017.
- Ayers EC, Li S, Medeiros LJ, et al.: Outcomes in patients with aggressive B-cell non-Hodgkin lymphoma after intensive frontline treatment failure. Cancer 126 (2): 293-303, 2020.
- Schuster SJ, Tam CS, Borchmann P, et al.: Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 22 (10): 1403-1415, 2021.
- Shah NN, Ahn KW, Litovich C, et al.: Is autologous transplant in relapsed DLBCL patients achieving only a PET+ PR appropriate in the CAR T-cell era? Blood 137 (10): 1416-1423, 2021.
- Shadman M, Pasquini M, Ahn KW, et al.: Autologous transplant vs chimeric antigen receptor T-cell therapy for relapsed DLBCL in partial remission. Blood 139 (9): 1330-1339, 2022.
- Cappell KM, Sherry RM, Yang JC, et al.: Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J Clin Oncol 38 (32): 3805-3815, 2020.
- Locke FL, Miklos DB, Jacobson CA, et al.: Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med 386 (7): 640-654, 2022.
- Elsawy M, Chavez JC, Avivi I, et al.: Patient-reported outcomes in ZUMA-7, a phase 3 study of axicabtagene ciloleucel in second-line large B-cell lymphoma. Blood 140 (21): 2248-2260, 2022.
- Kamdar M, Solomon SR, Arnason J, et al.: Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 399 (10343): 2294-2308, 2022.
- Bishop MR, Dickinson M, Purtill D, et al.: Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. N Engl J Med 386 (7): 629-639, 2022.
- Santomasso BD, Nastoupil LJ, Adkins S, et al.: Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J Clin Oncol 39 (35): 3978-3992, 2021.
- Shipp MA, Abeloff MD, Antman KH, et al.: International Consensus Conference on high-dose therapy with hematopoietic stem-cell transplantation in aggressive non-Hodgkin's lymphomas: report of the jury. Ann Oncol 10 (1): 13-9, 1999.
- Freedman AS, Takvorian T, Anderson KC, et al.: Autologous bone marrow transplantation in B-cell non-Hodgkin's lymphoma: very low treatment-related mortality in 100 patients in sensitive relapse. J Clin Oncol 8 (5): 784-91, 1990.
- Phillips GL, Fay JW, Herzig RH, et al.: The treatment of progressive non-Hodgkin's lymphoma with intensive chemoradiotherapy and autologous marrow transplantation. Blood 75 (4): 831-8, 1990.
- Chopra R, Goldstone AH, Pearce R, et al.: Autologous versus allogeneic bone marrow transplantation for non-Hodgkin's lymphoma: a case-controlled analysis of the European Bone Marrow Transplant Group Registry data. J Clin Oncol 10 (11): 1690-5, 1992.
- Ratanatharathorn V, Uberti J, Karanes C, et al.: Prospective comparative trial of autologous versus allogeneic bone marrow transplantation in patients with non-Hodgkin's lymphoma. Blood 84 (4): 1050-5, 1994.
- Mills W, Chopra R, McMillan A, et al.: BEAM chemotherapy and autologous bone marrow transplantation for patients with relapsed or refractory non-Hodgkin's lymphoma. J Clin Oncol 13 (3): 588-95, 1995.
- Philip T, Guglielmi C, Hagenbeek A, et al.: Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin's lymphoma. N Engl J Med 333 (23): 1540-5, 1995.
- Vellenga E, van Putten WL, van 't Veer MB, et al.: Rituximab improves the treatment results of DHAP-VIM-DHAP and ASCT in relapsed/progressive aggressive CD20+ NHL: a prospective randomized HOVON trial. Blood 111 (2): 537-43, 2008.
- Guglielmi C, Gomez F, Philip T, et al.: Time to relapse has prognostic value in patients with aggressive lymphoma enrolled onto the Parma trial. J Clin Oncol 16 (10): 3264-9, 1998.
- Vose JM, Anderson JR, Kessinger A, et al.: High-dose chemotherapy and autologous hematopoietic stem-cell transplantation for aggressive non-Hodgkin's lymphoma. J Clin Oncol 11 (10): 1846-51, 1993.
- Liberti G, Pearce R, Taghipour G, et al.: Comparison of peripheral blood stem-cell and autologous bone marrow transplantation for lymphoma patients: a case-controlled analysis of the EBMT Registry data. Lymphoma Working Party of the EBMT. Ann Oncol 5 (Suppl 2): 151-3, 1994.
- Vose JM, Zhang MJ, Rowlings PA, et al.: Autologous transplantation for diffuse aggressive non-Hodgkin's lymphoma in patients never achieving remission: a report from the Autologous Blood and Marrow Transplant Registry. J Clin Oncol 19 (2): 406-13, 2001.
- van Kampen RJ, Canals C, Schouten HC, et al.: Allogeneic stem-cell transplantation as salvage therapy for patients with diffuse large B-cell non-Hodgkin's lymphoma relapsing after an autologous stem-cell transplantation: an analysis of the European Group for Blood and Marrow Transplantation Registry. J Clin Oncol 29 (10): 1342-8, 2011.
- Freytes CO, Loberiza FR, Rizzo JD, et al.: Myeloablative allogeneic hematopoietic stem cell transplantation in patients who experience relapse after autologous stem cell transplantation for lymphoma: a report of the International Bone Marrow Transplant Registry. Blood 104 (12): 3797-803, 2004.
- Rezvani AR, Norasetthada L, Gooley T, et al.: Non-myeloablative allogeneic haematopoietic cell transplantation for relapsed diffuse large B-cell lymphoma: a multicentre experience. Br J Haematol 143 (3): 395-403, 2008.
- Ghosh N, Ahmed S, Ahn KW, et al.: Association of Reduced-Intensity Conditioning Regimens With Overall Survival Among Patients With Non-Hodgkin Lymphoma Undergoing Allogeneic Transplant. JAMA Oncol 6 (7): 1011-1018, 2020.
- Gisselbrecht C, Glass B, Mounier N, et al.: Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol 28 (27): 4184-90, 2010.
- Crump M, Kuruvilla J, Couban S, et al.: Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol 32 (31): 3490-6, 2014.
- Neelapu SS, Locke FL, Bartlett NL, et al.: Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377 (26): 2531-2544, 2017.
- Schuster SJ, Bishop MR, Tam CS, et al.: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380 (1): 45-56, 2019.
- Locke FL, Ghobadi A, Jacobson CA, et al.: Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 20 (1): 31-42, 2019.
- Abramson JS, Palomba ML, Gordon LI, et al.: Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396 (10254): 839-852, 2020.
- Lin JK, Muffly LS, Spinner MA, et al.: Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol 37 (24): 2105-2119, 2019.
- Jacobson CA, Hunter BD, Redd R, et al.: Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J Clin Oncol 38 (27): 3095-3106, 2020.
- Nastoupil LJ, Jain MD, Feng L, et al.: Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol 38 (27): 3119-3128, 2020.
- Salles G, Duell J, González Barca E, et al.: Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol 21 (7): 978-988, 2020.
- Zinzani PL, Pellegrini C, Gandolfi L, et al.: Combination of lenalidomide and rituximab in elderly patients with relapsed or refractory diffuse large B-cell lymphoma: a phase 2 trial. Clin Lymphoma Myeloma Leuk 11 (6): 462-6, 2011.
- Wiernik PH, Lossos IS, Tuscano JM, et al.: Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin's lymphoma. J Clin Oncol 26 (30): 4952-7, 2008.
- Sehn LH, Herrera AF, Flowers CR, et al.: Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol 38 (2): 155-165, 2020.
- Calabretta E, Hamadani M, Zinzani PL, et al.: The antibody-drug conjugate loncastuximab tesirine for the treatment of diffuse large B-cell lymphoma. Blood 140 (4): 303-308, 2022.
- Caimi PF, Ai W, Alderuccio JP, et al.: Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 22 (6): 790-800, 2021.
- Hamadani M, Radford J, Carlo-Stella C, et al.: Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 137 (19): 2634-2645, 2021.
- Dickinson MJ, Carlo-Stella C, Morschhauser F, et al.: Glofitamab for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 387 (24): 2220-2231, 2022.
- Lee AY, Connors JM, Klimo P, et al.: Late relapse in patients with diffuse large-cell lymphoma treated with MACOP-B. J Clin Oncol 15 (5): 1745-53, 1997.
- Haas RL, Poortmans P, de Jong D, et al.: Effective palliation by low dose local radiotherapy for recurrent and/or chemotherapy refractory non-follicular lymphoma patients. Eur J Cancer 41 (12): 1724-30, 2005.
Treatment of Lymphoblastic Lymphoma (LBL) / Acute Lymphocytic Leukemia (ALL)
LBL is a very aggressive form of non-Hodgkin lymphoma (NHL), which often occurs in young patients but not exclusively. LBL is the lymphomatous manifestation of ALL. The treatment paradigms are based on trials for ALL because LBL and ALL are considered different manifestations of the same biological disease. LBL is commonly associated with large mediastinal masses and has a high predilection for disseminating to bone marrow and the central nervous system (CNS). Intensive combination chemotherapy with CNS prophylaxis is the standard treatment of this aggressive histological type of NHL. Radiation therapy is sometimes given to areas of bulky tumor masses. Because these forms of NHL tend to progress quickly, combination chemotherapy is instituted rapidly once the diagnosis has been confirmed. For more information, see Adult Acute Lymphoblastic Leukemia Treatment.
The most important aspects of the pretreatment staging workup include careful review of the following pathological specimens:
- Bone marrow aspirate.
- Biopsy specimen.
- Cerebrospinal fluid cytology.
- Lymphocyte marker.
Treatment Options for LBL/ALL
Treatment options for LBL include the following:
- Intensive therapy.
- Radiation therapy.
New treatment approaches are being developed by the national cooperative groups. Other approaches include the use of bone marrow transplantation for consolidation.
For more information, see Adult Acute Lymphoblastic Leukemia Treatment.
Intensive therapy
Standard treatment is intensive combination chemotherapy with CNS prophylaxis.
Radiation therapy
Radiation therapy is sometimes given to areas of bulky tumor masses.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Treatment of Diffuse, Small Noncleaved-Cell / Burkitt Lymphoma
Diffuse, small noncleaved-cell/Burkitt lymphoma typically involves younger patients and represents the most common type of pediatric non-Hodgkin lymphoma (NHL).[1,2] High-grade B-cell lymphoma, not otherwise specified, includes lymphomas with Burkitt-like or blastoid morphology without double hit cytogenetics, and with germinal center B-cell phenotype.[3] Up to one-half of patients have a single MYC rearrangement. Optimal treatment is poorly defined because the diagnosis is rare. Burkitt lymphoma regimens with central nervous system (CNS) prophylaxis are usually chosen.[3]
Treatment Options for Diffuse, Small Noncleaved-Cell/Burkitt Lymphoma
Treatment options for diffuse, small, noncleaved-cell/Burkitt lymphoma include the following:
- Aggressive multidrug regimens.
- CNS prophylaxis.
Aggressive multidrug regimens
Treatment for diffuse, small noncleaved-cell/Burkitt lymphoma is usually an aggressive multidrug regimen similar to those used for the advanced-stage aggressive lymphomas (such as diffuse large cell).[4,5,6] Adverse prognostic factors include age older than 40 years, high serum lactate dehydrogenase (>3 times normal), Eastern Cooperative Oncology Group performance status of 2 or greater, and CNS involvement.[2] A retrospective review of 641 adult patients with Burkitt lymphoma from 30 U.S. cancer centers found a 3-year progression-free survival (PFS) rate of 64%. Nineteen percent of patients had CNS involvement, 14% had primary refractory disease, and the treatment-related mortality rate was 10%.[2]
Evidence (aggressive multidrug regimens):
- Aggressive combination chemotherapy patterned after that used in childhood Burkitt lymphoma has been very successful for adult patients. More than 60% of advanced-stage patients were free of disease at 5 years.[6,7,8,9]
- Rituximab has been incorporated into these aggressive combination chemotherapy regimens. A nonrandomized, single-arm, prospective, multicenter trial of 363 patients, aged 16 years to 85 years, showed a 5-year PFS rate of 71% and a 5-year overall survival rate of 80%.[5][Level of evidence C1]
CNS prophylaxis
Patients with diffuse, small noncleaved-cell/Burkitt lymphoma have a 20% to 30% lifetime risk of CNS involvement. CNS prophylaxis with methotrexate is recommended for all patients, usually given as four to six intrathecal injections.[10] For more information, see Adult Acute Lymphoblastic Leukemia Treatment.
Evidence (CNS prophylaxis):
- In a series of 41 patients treated with systemic and intrathecal chemotherapy, 44% of those who presented with CNS disease and 13% of those who relapsed with CNS involvement became long-term disease-free survivors.[11] CNS relapse patterns were similar whether or not patients received radiation therapy, but increased neurological deficits were noted among those patients who received radiation therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Blum KA, Lozanski G, Byrd JC: Adult Burkitt leukemia and lymphoma. Blood 104 (10): 3009-20, 2004.
- Evens AM, Danilov A, Jagadeesh D, et al.: Burkitt lymphoma in the modern era: real-world outcomes and prognostication across 30 US cancer centers. Blood 137 (3): 374-386, 2021.
- Olszewski AJ, Kurt H, Evens AM: Defining and treating high-grade B-cell lymphoma, NOS. Blood 140 (9): 943-954, 2022.
- Thomas DA, Faderl S, O'Brien S, et al.: Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 106 (7): 1569-80, 2006.
- Hoelzer D, Walewski J, Döhner H, et al.: Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood 124 (26): 3870-9, 2014.
- Roschewski M, Dunleavy K, Abramson JS, et al.: Multicenter Study of Risk-Adapted Therapy With Dose-Adjusted EPOCH-R in Adults With Untreated Burkitt Lymphoma. J Clin Oncol 38 (22): 2519-2529, 2020.
- Magrath I, Adde M, Shad A, et al.: Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol 14 (3): 925-34, 1996.
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
- Mead GM, Sydes MR, Walewski J, et al.: An international evaluation of CODOX-M and CODOX-M alternating with IVAC in adult Burkitt's lymphoma: results of United Kingdom Lymphoma Group LY06 study. Ann Oncol 13 (8): 1264-74, 2002.
- Rizzieri DA, Johnson JL, Niedzwiecki D, et al.: Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: final results of Cancer and Leukemia Group B Study 9251. Cancer 100 (7): 1438-48, 2004.
- Magrath IT, Haddy TB, Adde MA: Treatment of patients with high grade non-Hodgkin's lymphomas and central nervous system involvement: is radiation an essential component of therapy? Leuk Lymphoma 21 (1-2): 99-105, 1996.
Treatment of NHL During Pregnancy
General Information About NHL During Pregnancy
Non-Hodgkin lymphomas (NHL) occur more frequently than Hodgkin lymphoma in an older population. This age difference may account for fewer reports of NHL in pregnant patients.[1]
Stage Information for NHL During Pregnancy
To avoid exposure to ionizing radiation, magnetic resonance imaging is the preferred tool for staging evaluation.[2] For more information, see the Stage Information for NHL section.
Treatment Option Overview for NHL During Pregnancy
Table 5. Treatment Options for Non-Hodgkin Lymphoma (NHL) During Pregnancy| Stage | Treatment Options |
|---|
| Indolent NHL During Pregnancy | Delay treatment until after delivery |
| Aggressive NHL During Pregnancy | Immediate therapy |
| Early delivery, when feasible |
| Termination of pregnancy |
Indolent NHL During Pregnancy
Treatment may be delayed for those women with an indolent NHL.
Aggressive NHL During Pregnancy
Immediate therapy
According to anecdotal case series, most NHLs in pregnant patients are aggressive, and delay of therapy until after delivery appears to have poor outcomes.[1,3,4,5] Consequently, some investigators favor immediate therapy, even during pregnancy.[5] In a review of 121 patient case reports from 74 papers, one-half of the patients had very aggressive lymphomas, such as Burkitt lymphoma, and one-half of the patients had involvement of the breast, ovaries, uterus, or placenta.[6] One-half of the patients received therapy antepartum, and the 6-month survival rate was reported at 53%, with a live-birth rate of 83%.[6][Level of evidence C3]
A multicenter retrospective analysis of 50 patients described pregnancy termination in 3 patients, deferral of therapy to postpartum in 15 patients (median 30 weeks gestation), and antenatal therapy applied to the remaining 32 patients (median 21 weeks gestation, all done after the first trimester).[7] With a median follow-up of 41 months, the 3-year progression-free survival rate was 53%, and the overall survival rate was 82%, using R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) or modifications of this regimen.[7][Level of evidence C3]
Early delivery when feasible
For some women, early delivery, when feasible, may minimize or avoid exposure to chemotherapy or radiation therapy.
Termination of pregnancy
Termination of pregnancy in the first trimester may be an option that allows immediate therapy for women with aggressive NHL.
Evidence (treatment effect on children exposed in utero):
- With follow-up ranging from several months to 11 years, children who were exposed to high-dose doxorubicin-containing combination chemotherapy in utero (especially during the second and third trimester) had normal growth and no evidence of congenital malformations.[5,8,9,10] For most of the chemotherapeutic agents used for the treatment of NHL, there are no data regarding long-term effects on children exposed in utero.
- In one anecdotal case, a newborn exposed to a rituximab-containing regimen in utero was born with no circulating B lymphocytes. The newborn was otherwise healthy and recovered the circulating B lymphocytes by age 6 months with no unusual or persisting intercurrent infections.[11]
References:
- Ward FT, Weiss RB: Lymphoma and pregnancy. Semin Oncol 16 (5): 397-409, 1989.
- Nicklas AH, Baker ME: Imaging strategies in the pregnant cancer patient. Semin Oncol 27 (6): 623-32, 2000.
- Steiner-Salz D, Yahalom J, Samuelov A, et al.: Non-Hodgkin's lymphoma associated with pregnancy. A report of six cases, with a review of the literature. Cancer 56 (8): 2087-91, 1985.
- Spitzer M, Citron M, Ilardi CF, et al.: Non-Hodgkin's lymphoma during pregnancy. Gynecol Oncol 43 (3): 309-12, 1991.
- Gelb AB, van de Rijn M, Warnke RA, et al.: Pregnancy-associated lymphomas. A clinicopathologic study. Cancer 78 (2): 304-10, 1996.
- Horowitz NA, Benyamini N, Wohlfart K, et al.: Reproductive organ involvement in non-Hodgkin lymphoma during pregnancy: a systematic review. Lancet Oncol 14 (7): e275-82, 2013.
- Evens AM, Advani R, Press OW, et al.: Lymphoma occurring during pregnancy: antenatal therapy, complications, and maternal survival in a multicenter analysis. J Clin Oncol 31 (32): 4132-9, 2013.
- Avilés A, Díaz-Maqueo JC, Torras V, et al.: Non-Hodgkin's lymphomas and pregnancy: presentation of 16 cases. Gynecol Oncol 37 (3): 335-7, 1990.
- Moore DT, Taslimi MM: Multi-agent chemotherapy in a case of non-Hodgkin's lymphoma in second trimester of pregnancy. J Tenn Med Assoc 84 (9): 435-6, 1991.
- Nantel S, Parboosingh J, Poon MC: Treatment of an aggressive non-Hodgkin's lymphoma during pregnancy with MACOP-B chemotherapy. Med Pediatr Oncol 18 (2): 143-5, 1990.
- Mandal PK, Dolai TK, Bagchi B, et al.: B cell suppression in newborn following treatment of pregnant diffuse large B-cell lymphoma patient with rituximab containing regimen. Indian J Pediatr 81 (10): 1092-4, 2014.
Changes to This Summary (05 / 18 / 2023)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Indolent Non-Hodgkin Lymphoma (NHL]
Added text to state that CD19-directed chimeric antigen receptor (CAR) T cells may be used in patients who have disease progression after two or more prior lines of therapy (cited Jacobson et al. as reference 30). Also added text to state that mosunetuzumab may also be used in this setting (cited Bartlett et al. as reference 31).
Added Gertz as reference 48. Also revised text to state that symptomatic patients with a serum viscosity of four or lower are usually started directly on chemoimmunotherapy or biologically directed therapies. Therapy may be required to correct hemolytic anemia in patients with chronic cold agglutinin disease; rituximab, bendamustine, and steroids are often used. Sutimlimab can reduce hemolysis when therapies directed at the lymphoplasmacytic lymphoma prove ineffective (cited Röth et al. as reference 49).
Added text to state that splenic marginal zone lymphoma is a distinct clinical entity that usually presents with massive splenomegaly. A variant form of mucosa-associated lymphatic tissue lymphoma is known as immunoproliferative small intestinal disease (cited Rossi et al. as reference 80).
Revised text to state that among patients with concomitant hepatitis C virus (HCV) infection, 40% to 60% attained a complete or partial remission after loss of detectable HCV RNA with antiviral treatment (cited Merli et al. as reference 119).
Aggressive NHL
Revised text to state that, among patients with advanced-stage disease, 50% are cured with doxorubicin-based combination chemotherapy and rituximab, typically R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
Added text to state that true anaplastic lymphoma kinase (ALK)–positive large B-cell lymphomas are extremely rare, and they do not respond well to conventional R-CHOP therapy. Anecdotal responses to ALK inhibitors like lorlatinib or alectinib have been reported (cited Soumerai et al. as reference 22 and level of evidence C3).
Added Savage as reference 37.
Added text to state that among patients with primary mediastinal large B-cell lymphoma who had received two prior lines of therapy, more than one-half of patients who received CAR T-cell therapy with lisocabtagene maraleucel had disease response (cited Abramson et al. as reference 53 and level of evidence C3).
Added text to state that among patients with follicular large cell lymphoma who had received two prior lines of therapy, more than one-half of patients who received CAR T-cell therapy with lisocabtagene maraleucel had disease response.
Added text to state that a retrospective review of 84 patients with ALK-negative anaplastic large cell lymphoma (ALCL) suggested a survival benefit with autologous stem cell transplantation. This hypothesis requires confirmation in a randomized prospective trial (cited Brink et al. as reference 72).
Added text to state that primary cutaneous ALCL is a distinct entity that is typically ALK-negative and has a very indolent/low-grade clinical course.
Added text about a phase II trial of 131 previously untreated patients with mantle cell lymphoma (MCL) aged 65 years or younger. One year of ibrutinib plus 4 weeks of rituximab resulted in a complete response (CR) rate of 89% prior to any chemotherapy consolidation (cited 2022 Wang et al. as reference 210 and level of evidence C3). Another phase II trial using ibrutinib plus rituximab included asymptomatic patients with previously untreated MCL; the CR rate was 87% (cited Giné et al. as reference 211 and level of evidence C3).
Added Hermine et al. as reference 220.
Revised text about the results of phase II trial of brexucabtagene autoleucel in patients with relapsed or refractory MCL whose disease did not respond to ibrutinib or acalabrutinib (cited 2023 Wang et al. as reference 232).
Stage Information for NHL
Added text to state that in a retrospective review of over 32,000 cases of lymphoma, up to 40% of diagnoses were made by core needle biopsy, and 60% were made by excisional biopsy (cited Syrykh et al. as reference 1). Core needle biopsy provided a definite diagnosis in 92.3% of cases, and excisional biopsy provided a definite diagnosis in 98.1% of cases.
Added text about retrospective study of 580 patients with follicular lymphoma that showed no improvement in assessing response to therapy when bone marrow biopsy was added to radiological imaging (cited Rutherford et al. as reference 10).
Treatment of Indolent, Noncontiguous Stage II/III/IV NHL
Added Morschhauser et al. as reference 59.
Revised text about the results of the RELEVANCE trial, which included 1,030 patients with previously untreated follicular lymphoma who were treated with rituximab plus lenalidomide for 18 months or rituximab plus chemotherapy (usually R-CHOP).
Treatment of Indolent, Recurrent NHL
Revised the list of treatment options for indolent, recurrent NHL to include bispecific T-cell engagers.
Revised text to state that CAR T-cell therapy, with the autologous anti-CD19 therapeutics axicabtagene ciloleucel, lisocabtagene maraleucel, or tisagenlecleucel, has been approved for patients with relapsed follicular lymphoma after two or more lines of prior therapy.
Revised results of a phase II trial of axicabtagene ciloleucel.
Added text about a phase II trial of 98 patients with relapsed or refractory follicular lymphoma after two or more lines of prior therapy who received anti-CD19 CAR T-cell therapy with tisagenlecleucel (cited Fowler et al. as reference 33 and level of evidence C3).
Added Bispecific T-cell engagers as a new subsection.
Treatment of Aggressive Stage I and Aggressive, Contiguous Stage II NHL
Added text about a retrospective analysis of 341 patients with stage I disease who received R-CHOP (or a similar regimen) with or without radiation therapy (cited Bobillo et al. as reference 7 and level of evidence C3).
Added text to state that although a retrospective study suggested that patients with stage I extranodal disease and a positive positron emission tomography scan at the end of therapy may benefit from radiation therapy, this hypothesis must be confirmed in a prospective randomized trial.
Treatment of Aggressive, Noncontiguous Stage II/III/IV NHL
Added Nowakowski et al. as reference 10.
Treatment of Aggressive, Recurrent NHL
Revised the list of treatment options for aggressive, recurrent NHL to include bispecific T-cell engagers and palliative radiation therapy.
Revised text about the results of a prospective randomized trial of CAR T-cell therapy to state that clinically meaningful and statistically significant differences in quality of life were obtained in the CAR T-cell arm at day 100 and day 150, compared with the standard of care (cited Elsawy et al. as reference 8 and level of evidence A2).
Revised text about the results of another prospective randomized trial of CAR T-cell therapy to state that grade 3 cytokine release syndrome occurred in 1% of patients, and grade 3 neurotoxicity occurred in 4% of patients.
Added Calabretta et al. as reference 41.
Added Bispecific T-cell engagers as a new subsection.
Treatment of Diffuse, Small Noncleaved-Cell/Burkitt Lymphoma
Added text to describe high-grade B-cell lymphoma, not otherwise specified (cited Evens et al as reference 2 and Olszewski et al. as reference 3).
Added text about adverse prognostic factors for diffuse, small noncleaved-cell/Burkitt lymphoma and about the results of a retrospective review of 641 adult patients with Burkitt lymphoma.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult non-Hodgkin lymphoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Non-Hodgkin Lymphoma Treatment are:
- Eric J. Seifter, MD (Johns Hopkins University)
- Cole H. Sterling, MD (Johns Hopkins)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Non-Hodgkin Lymphoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/lymphoma/hp/adult-nhl-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389492]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2023-05-18